Introduction
Hepatocellular carcinoma (HCC) is the third leading
cause of cancer-related deaths worldwide (1). Fibrolamller hepatocellular carcinoma
(FL-HCC) is a rare subtype of HCC, which typically occurs in
adolescents and young adults (2–4). FL-HCC
has unique histological characteristics such as large, polygonal
tumor cells with eosinophilic or oncocytic cytoplasm, large nuclei
with prominent single nucleolus, and pericellular lamellar collagen
fibrosis (5,6) that sets it apart from conventional HCC.
In addition, patients with FL-HCC generally do not have underlying
liver disease (7) as opposed to
conventional HCC which is commonly associated with cirrhosis and/or
chronic viral hepatitis (8). Surgical
resection is the predominant mode of treatment for FL-HCC and
provides the only chance at a cure (9). However, the mortality rates remain high
as a significant percentage of patients present with advanced or
unresectable disease. Even patients who undergo surgical resection
have a high risk of tumor recurrence. Therefore, overall survival
of patients with FL-HCC will continue to be poor until effective
chemotherapeutic agents are identified.
A recent approach to treating cancers has focused on
activating the tumor suppressor function of p53, known as the
‘guardian of the genome’ (10,11), which
under normal conditions undergoes activation once there is DNA
damage or instability (12).
Ataxia-telangiectasia mutated (ATM), is a serine/threonine kinase
that is often initially activated upon DNA damage (13,14) and
subsequently signals p53 to perform DNA repair, inhibit abnormal
cell growth, or induce apoptosis (15–18).
Conversely, p53 activity may be negatively regulated by mouse
double minute 2 homolog (MDM2) or mouse double minute 4, human
homolog (MDM4) (19). MDM2, an E3
ubiquitin-protein ligase, leads to the degradation of p53 through
the proteasomal pathway and MDM4 exerts its inhibitory activity via
binding to p53 transactivation domain (20–23).
Approximately 50% of all human cancers contain p53
mutations, which in turn results in the loss of p53 tumor
suppressor function leading to cancer initiation and progression
(24). There is now increasing
evidence that even in cancers with non-mutated p53, the function of
p53 is altered such that it favors the biology of the cancer cell
and prevents the apoptosis of the transformed cell. One mechanism
responsible for the dysregulation of p53 function has been
attributed to the inhibitory effect of MDM4 (25,26). The
overexpression of MDM4 has been reported in various cancer types
with non-mutated p53 (27). For
example, MDM4 expression is elevated in 65% of human melanomas and
in an animal model with Nras oncogene activation;
overexpression of MDM4 increased cancer promotion in melanocytes
(28). Similarly, in vitro
studies of breast cancer lacking p53 mutations demonstrated that
MDM4 knockdown inhibited cell growth (29). Collectively, these studies indicated
that overexpression of MDM4 may result in dysregulation of p53
function and inhibiton of MDM4 may provide a therapeutic approach
for treating cancers that lack p53 mutations.
As opposed to conventional HCC, which is commonly
associated with mutations in p53 (8,30), FL-HCC
is not linked to any p53 mutations (24,31,32),
however, there is evidence for p53 pathway dysregulation based on
transcriptome analysis of FL-HCC (33). Additionally, studies have reported
that conventional HCC tumors lacking p53 mutations have increased
MDM4 expression indicating that p53 function is disrupted (1,31,34,35). Thus,
in the present study, it was examined whether this tumor type is
also associated with p53 dysregulation by examining the DDR
components; ATM, p53, and MDM4.
Materials and methods
Tumor samples
Seven FL-HCC tumor samples and their matched
non-neoplastic liver samples were obtained from patients who
underwent hepatic resection or liver transplantation under an
approved protocol by the Institutional Review Board (IRB) at Boston
Children's Hospital. Written informed consent was obtained from the
parents of patients under 18 years of age. The neoplastic and
non-neoplastic tissues were reviewed by an expert liver pathologist
(ARPA).
Histology and
immunohistochemistry
Samples were fixed, embedded in paraffin, sectioned,
and stained using conventional histological techniques by the Core
Histology Facility at Boston Children's Hospital. Hematoxylin and
eosin (H&E) staining was performed on 4-µm tissue sections as
previously reported by LaQuaglia et al (36). Briefly, the paraffin slides were
dehydrated for 60 sec in methanol, followed by 120 sec staining in
Harris' Hematoxylin (Sigma Aldrich; Merck KGaA). The slides were
washed in water for 20 sec. Next, the slides were incubated in the
bluing reagent for 60 sec (Thermo Fisher Scientific, Inc.) followed
by 30-sec washes in water and 95% ethanol. The slides were further
incubated in Eosin Yellow (Thermo Fisher Scientific, Inc.) for 60
sec followed by a dehydration process in a series of ethanol and
xylene. In order to perform immunohistochemistry (IHC), tissues
were deparaffinized and retrieved with antigen unmasking solution
(Vector Laboratories, Inc.). The sections were incubated in
hydrogen peroxide (3%) for 5 min for blocking the endogenous
peroxidase activity. The samples were then blocked for 2 h at room
termperature (RT) using normal goat serum-based reagent. Tissue
sections were incubated overnight at 4°C using the following
primary antibodies: MDMX (cat. no. NBP1-28862; dilution 1:100;
Novus Biologicals, LLC), p53 (cat. no. PAb1801, dilution 1:100,
Abcam) p-p53-S15 (cat. n. 9284S, dilution 1:150; Cell Signaling
Technologies, Inc.), ATM (dilution 1:100; cat. no. NB100-104; Novus
Biologicals, LLC), p-ATM-Ser1981 (cat. no. sc-47739; dilution 1:50;
Santa Cruz Biotechnology, Inc.). The tissue sections were incubated
in prediluted biotinylated anti-mouse or anti-rabbit secondary
antibodies (cat. no. PK-8200; Vector Laboratories) for 45 min at
room temperature followed by 1-h incubation in Vecta Stain R.T.U
(Vector Laboratories, Inc.) at room temperature. Vector reagents
and DAB (diamonobenzidine) peroxidase substrate (Vector
Laboratories, Inc.) were applied to detect the antibody
staining.
Immunoblots, quantification and
statistics
Tumor and non-neoplastic tissue samples were
dissociated and lysed in ice-cold RIPA buffer supplemented with
protease inhibitors, phosphatase inhibitors, using a Tissue Tearor
Homogenizer (Biospec Products, Inc). Whole cell protein lysates
(15–30 µg) containing Lamelli buffer plus β-mercaptoethanol, were
heated at 95°C for 10 min. The samples were then resolved in 4–20%
precast Tris-glycine gels (Invitrogen; Thermo Fisher Scientific,
Inc.) and transferred onto PVDF membranes using Trans-Blot Turbo
Transfer System (both from Bio-Rad Laboratories, Inc.). Membranes
were subjected to blocking for 2 h at room termperature in 5%
non-fat dry milk in tris-buffered saline and 0.1% Tween-20.
Immunoblot membranes were incubated in primary antibodies at 4°C
overnight against the following antibodies: MDM4 (Abgent, Inc.;
cat. no. ALS13152, dilution-1:1,000), p53 (Abcam; cat. no. PAb1801,
dilution 1:1,000), phospho p53-S15 (Cell Signaling Technologies,
Inc., cat. no. 9284S; dilution 1:1,000), PKA (Santa Cruz
Biotechnology, Inc., cat. no. sc-903; dilution 1:1,000), α-tubulin
(GeneTex, Inc.; cat. no. GTX628802; dilution 1:3,000), GAPDH (Cell
Signaling Technologies, Inc., cat. no. 97166; dilution 1:3,000 and
GenTex, Inc., cat. no. GTX627408; dilution 1:3,000) and β-actin
(Cell Signaling Technologies, Inc.; cat. no. 4970S; dilution
1:3,000). This was followed by three 10-min washes. Membranes were
then incubated in horseradish peroxidase (HRP) conjugated secondary
antibodies (Jackson ImmunoResearch Laboratories, INC, anti-mouse-
cat. no. 205-005-108; dilution 1:5,000, anti-rabbit; cat. no.
111-035-144; dilution 1:5,000) at room temperature for 45 min
followed by three washes with TBST. Immunoblots were visualized
using an enhanced chemiluminescence (ECL) kit (Bio-Rad
Laboratories, Inc. and Thermo Fisher Scientific, Inc.). We did not
have ample tumor tissue from patient P6 to perform all of the
western blot analyses.
Protein quantification was performed using ImageJ
software version:1.48v (National Institutes of Health) (normalized
to GAPDH or β-actin). GraphPad Prism 7 (GraphPad Software) was used
to generate graphs and Mann Whitney U test was used to generate
P-values.
RNA expression analysis
Total cellular RNA from tumor and non-neoplastic
liver samples was extracted using the E.Z.N.A midi kit (Qiagen,
Inc.). One microgram of RNA was subjected to reverse transcription
using the iScript cDNA synthesis kit (Bio-Rad Laboratories, Inc.).
Gene amplification was performed using SYBR Rox and an AB
7000 Real-Time PCR system. Thermocycling conditions used were: 37
cycles at 95°C for 30 sec, 59°C for 1 min, and 72°C for 30 sec with
the following primers MDM4 (Hs00910358_s1) and GAPDH
(Hs02758991_g1) (all from Applied Biosystems; Thermo Fisher
Scientific, Inc.). Each gene reaction was performed in duplicates
and all transcript values were normalized to GAPDH. The Cq
values for each gene was generated using the AB7000 software and
data analysis was performed using the 2−ΔΔCq method. The
values were first normalized to GAPDH. Then to generate the
final folds, the values were renormalized to the non-neoplastic
samples.
Microscopy, semiquantative scoring
system and statistical analysis
All images were obtained using an EVOS Imaging
System (Invitrogen; Thermo Fisher Scientific, Inc.). In the present
analysis for IHC, 5/7 tumor tissues were compared to non-neoplastic
tissue from the same patient. The immunoreactive score (IRS) was
utilized to evaluate each immunohistochemical stain and performed
by a single pathologist (JP) (37).
Histologic evaluation included the percentage of positive cells in
the lesional and non-lesional areas and the staining intensity. IRS
was a product of multiplication between the positive-cell
proportion score (0–4) and staining intensity score (0–3). The
final score was interpreted as negative (0–1), mild (2–3), moderate
(4–8)
or strongly positive (9–12). A detailed calculation for generating
the IRS is illustrated in Table SI.
Mann-Whitney test was performed to compare the median data values
between non-neoplastic and neoplastic cells. The non-parametric
two-sided Mann-Whitney test was conducted using Graphpad Prism 7
software for all statistical analyses and a P-value <0.05 was
considered statistically significant.
Results
Patient characteristics and
histopathology
All patients in the present study, were male and the
median age of the patients in the present study was 13 years.
Previous studies have reported that FL-HCC occurs predominantly in
females (2), however, others have
reported that both males and females are equally likely to be
affected (38,39). Two patients (P3 and P6) presented to
our institution with metastatic disease. Patient P6 had prior liver
resection at an outside institution and presented with recurrent
metastatic disease and underwent tumor biopsy. The median follow-up
for the entire cohort was 26 months (range 16–70 months). All
patients except P7 had undergone neo-adjuvent therapy prior to
surgery. Patient demographics, tumor data, and clinical course is
summarized in Table I. Tumor and
non-neoplastic livers from 4 patients were collected following
hepatic resection; 2 after transplantation; and 1 following
core-needle biopsy of a recurrent tumor. All tumors were reviewed
by an expert liver pathologist (ARPA) and revealed to be
characteristic of FL-HCC and all demonstrated microvascular
invasion. All but one patient (P2) had tumor recurrence following
surgery, at a median time of 155 days (range 0–563 days). Two
patients succumbed to this disease since surgery, both due to tumor
progression. Overall survival at 5 years was 71%.
 | Table I.Patient and tumor
characteristics. |
Table I.
Patient and tumor
characteristics.
| Patient | Age at
diagnosis | Sex | BMI | AFP (ng/ml) | Tumor size
(cm) | Extent of
tumor | Surgery | Clinical
course | Date of
recruitmenta |
|---|
| P1 | 13.7 | M | 19.6 | 1.1 | 19.5×13.4×8.5 | Liver segments
II/III, IVa/IVb; tumor thrombus in PV, HV, IVC and RA | Total hepatectomy
on cardiopulmonary bypass; ex vivo left hepatectomy; right
lobe autotransplantation | Underwent resection
of brain metastasis at 19 months post-liver resection; alive at
75-month follow-up | 02/26/2013 |
| P2 | 13.6 | M | 14.4 | 1 | 9×8×6.5 | Liver left lobe;
1.1 cm lesion in right lobe; tumor thrombus in PV, SV, SMV | Multivisceral
transplant (liver, intestine, pancreas, spleen, stomach) | Alive at 77-month
follow-up after transplant with no evidence of disease | 07/16/2013 |
| P3 | 13.4 | M | 16.5 | 328897 | 14.1×12×7.5 | Liver right lobe;
hepatic vein | Right
trisegmentectomy | Alive at 55 months
with metastatic disease | 11/06/2014 |
| P4 | 11.4 | M | 14.1 | 5 | 8.6×6.1×6.0 | Liver segments
II/IVa | Left
hepatectomy | Recurrence at 108
days; died of recurrence 26 months after initial diagnosis | 10/27/2014 |
| P5 | 15.8 | M | 31.3 | 148 | 18.5×9×8 | Right lobe; portal
vein invasion | Right
trisegmentectomy | Recurrence at 9
months; salvage transplant at 23 months; alive with recurrence at
45 months after initial diagnosis | 09/12/2015 |
| P6 | 10.8 | M | N/A | 4 | 10.0×6.8×5.5 | Caudate lobe
recurrence; involvement of PV; HA; and CBD | Attempted
resection; core needle biopsy | Recurrence at 64
months; died 87 months after initial diagnosis | 09/22/2016 |
| P7 | 14 | M | 16 | 14 | 10.8×2.3×8.3 | Liver segments
II/III/IV, invasion of CBD | Liver
transplant | Recurrence at 279
days after transplant; alive at 15 months post-transplant | 12/22/2017 |
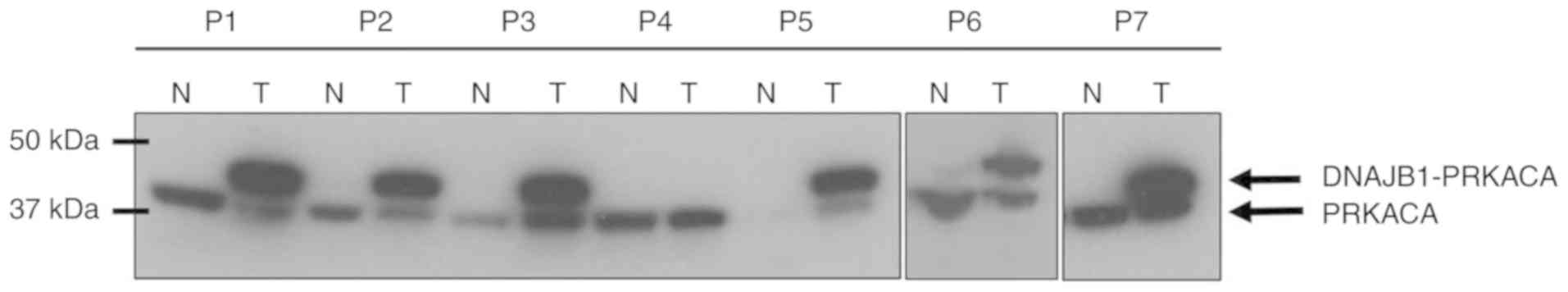
PRKACA-DNAJB1 fusion protein is
expressed in the majority of FL-HCC
A recent study described the presence of
PRKACA-DNAJB1 fusion gene in chromosome 19 in FL-HCC tumors
(40). This fusion gene results from
a chromosomal deletion of 400 kilobase pairs, which generates a
fusion chimera between exon 1 of DNAJB1 (Heat Shock Protein 40) and
exons 2 through 10 of the catalytic subunit of protein kinase A,
PRKACA (40,41). Using western blot analysis, the
presence of PRKACA-DNAJB1 fusion protein with an antibody targeting
the c-terminus of PRKACA was assessed. It was revealed that 6/7
tumor samples expressed the fusion protein (Fig. 1). The native PRKACA band was detected
at ~41 kDa and the band shift at ~46 kDa in the immunoblots
represented the presence of the fusion protein. Tumor sample from
patient P4 was confirmed as FL-HCC histologically, however, the
fusion protein was not present in this tumor.
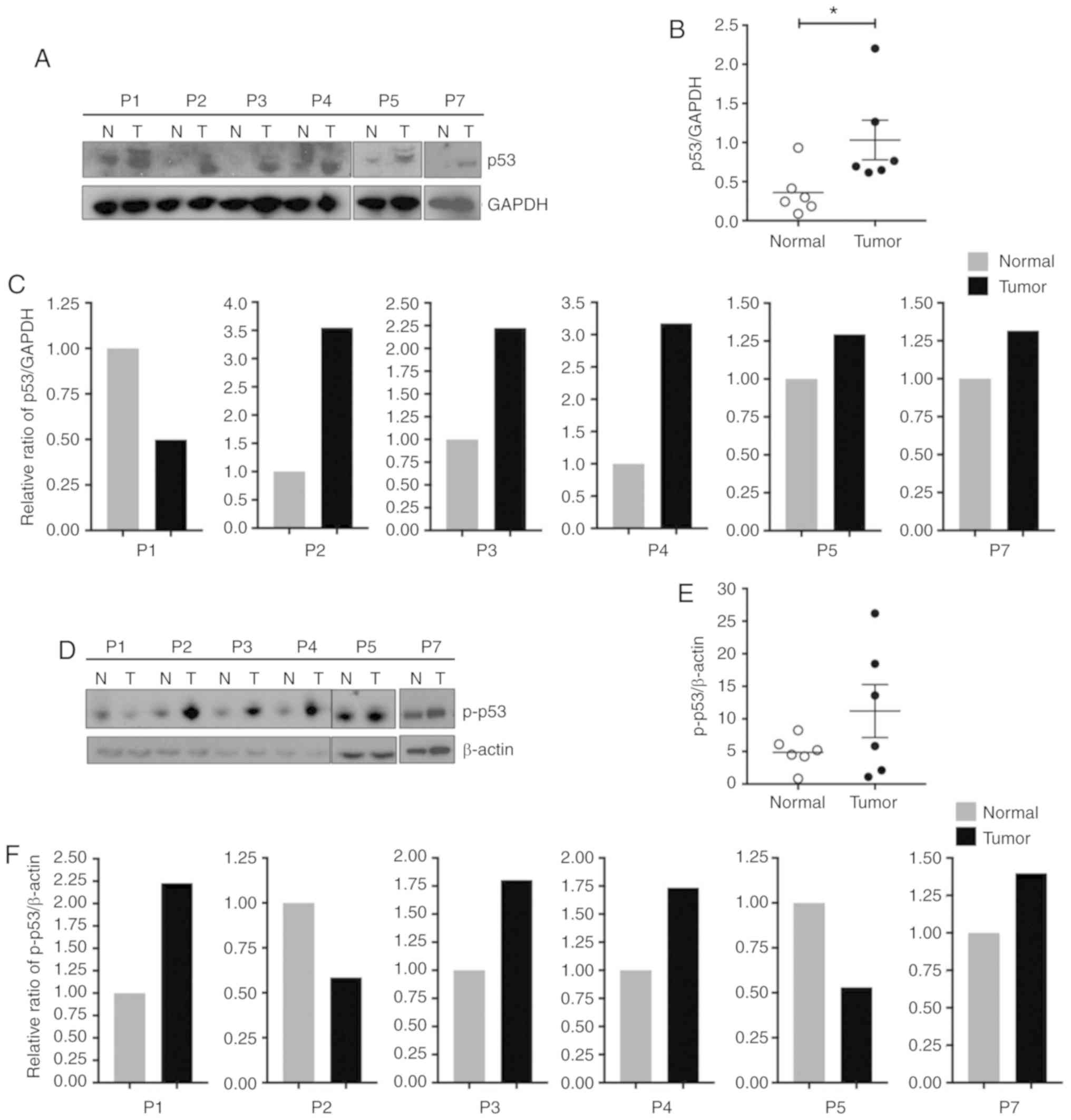
p53 protein expression is increased in
FL-HCC
The expression of p53 and its activated form,
phosphorylated p53 (p-p53) in tumor tissue was then determined.
Western blot analysis was performed from whole cell protein lysates
using p53 and p-p53 (serine 15) antibodies and the corresponding
protein levels were assessed. Quantitative analysis of the
immunoblots revealed that p53 protein was significantly increased
in tumor tissues compared to matching non-neoplastic liver tissues
(Mann-Whitney test, P=0.03, Fig. 2A and
B). As revealed in Fig. 2C,
individual immunoblot quantification demonstrated that p53 was
increased in 5/6 tumor samples. Furthermore, western blot analysis
revealed that although the overall p-p53 expression was not
significantly elevated (Mann-Whitney test, P=0.39), there was an
increase in p-p53 in 4/6 tumors (Fig.
2D-F).
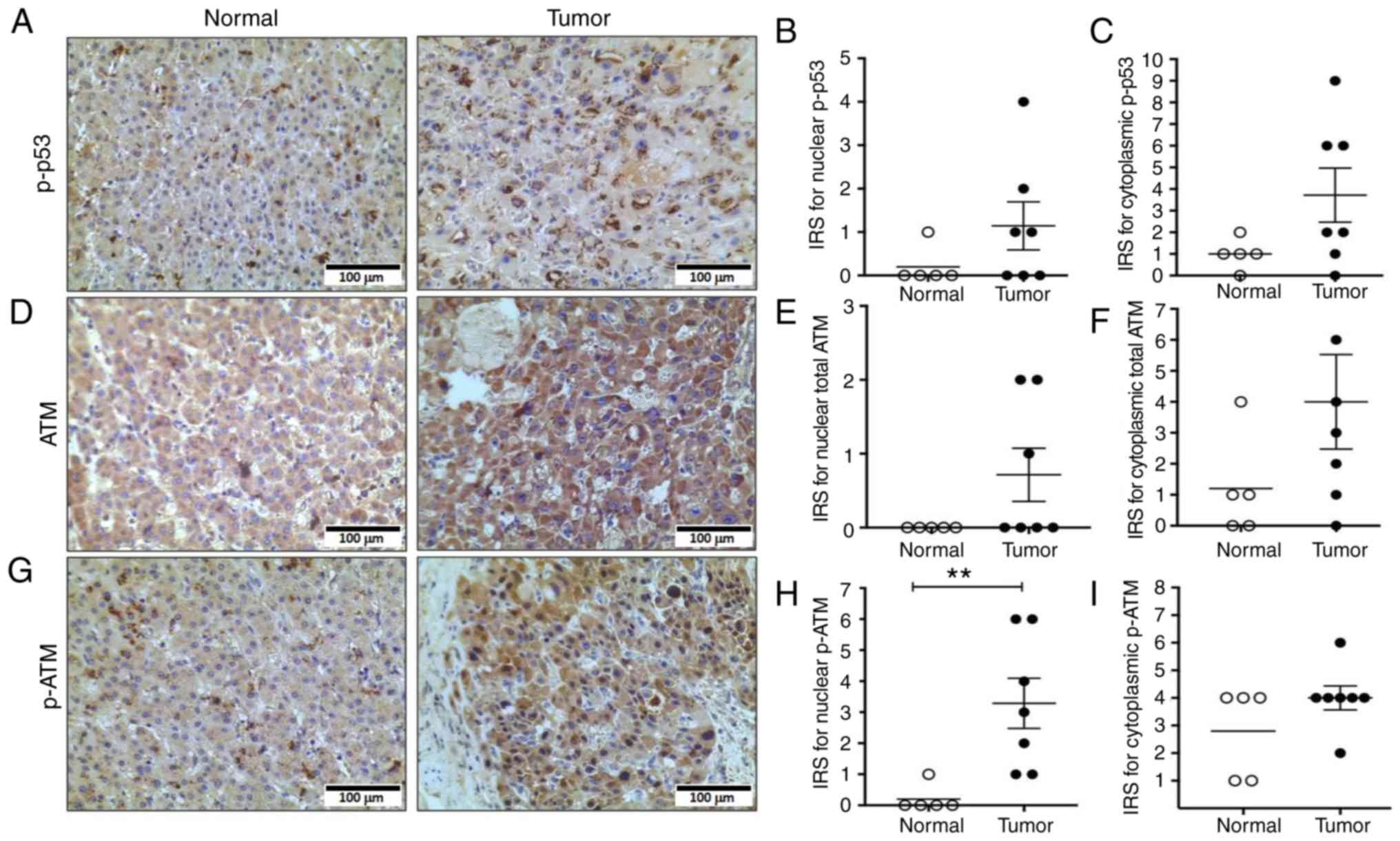
Next, the subcellular localization pattern of p53
and p-p53 was assessed. IHC was performed on paraffin-embedded
sections using anti-p53 and anti-p-p53 antibodies followed by
nuclear and cytoplasmic assessment of each protein using the IRS
quantification system. There was very low nuclear and cytoplasmic
p53 staining in both tumor and normal liver tissue (Fig. S1). In contrast, p-p53 was detected in
both the nucleus and cytoplasm of tumor cells with no overall
significant differences between tumor cells and non-neoplastic
cells (P=0.28 for nuclear p-p53 and P=0.14 for cytoplasmic p-p53;
Mann-Whitney test, Fig. 3A-C).
However, the majority of the patients had higher IRS values for
cytoplasmic p-p53 in the tumor cells compared to the non-neoplastic
cells.
ATM, a p53 regulator, is activated in
FL-HCC
Next, the expression and localization of ATM protein
kinase, a known upstream regulator of p53, was examined (42). The detection of ATM and
phosphorylated-ATM (serine 1981; p-ATM) protein levels was not
feasible due to unsuitable antibodies for western blotting.
However, protein expression and localization of ATM and p-ATM in
tumors cells and non-neoplastic cells were examined using IHC
(Fig. 3D and G) and IRS values were
generated for nuclear and cytoplasmic localization of total ATM and
p-ATM proteins (Fig. 3E and F and H and
I, respectively). There was no statistically significant
difference in nuclear or cytoplasmic ATM expression between tumor
cells and non-neoplastic cells (P=0.20 for nuclear ATM and P=0.16
for cytoplasmic ATM; Mann-Whitney test). However, there was a
significant increase in p-ATM levels in the nuclei of tumors
compared to normal cells (Mann-Whitney test, P=0.005, Fig. 3H).
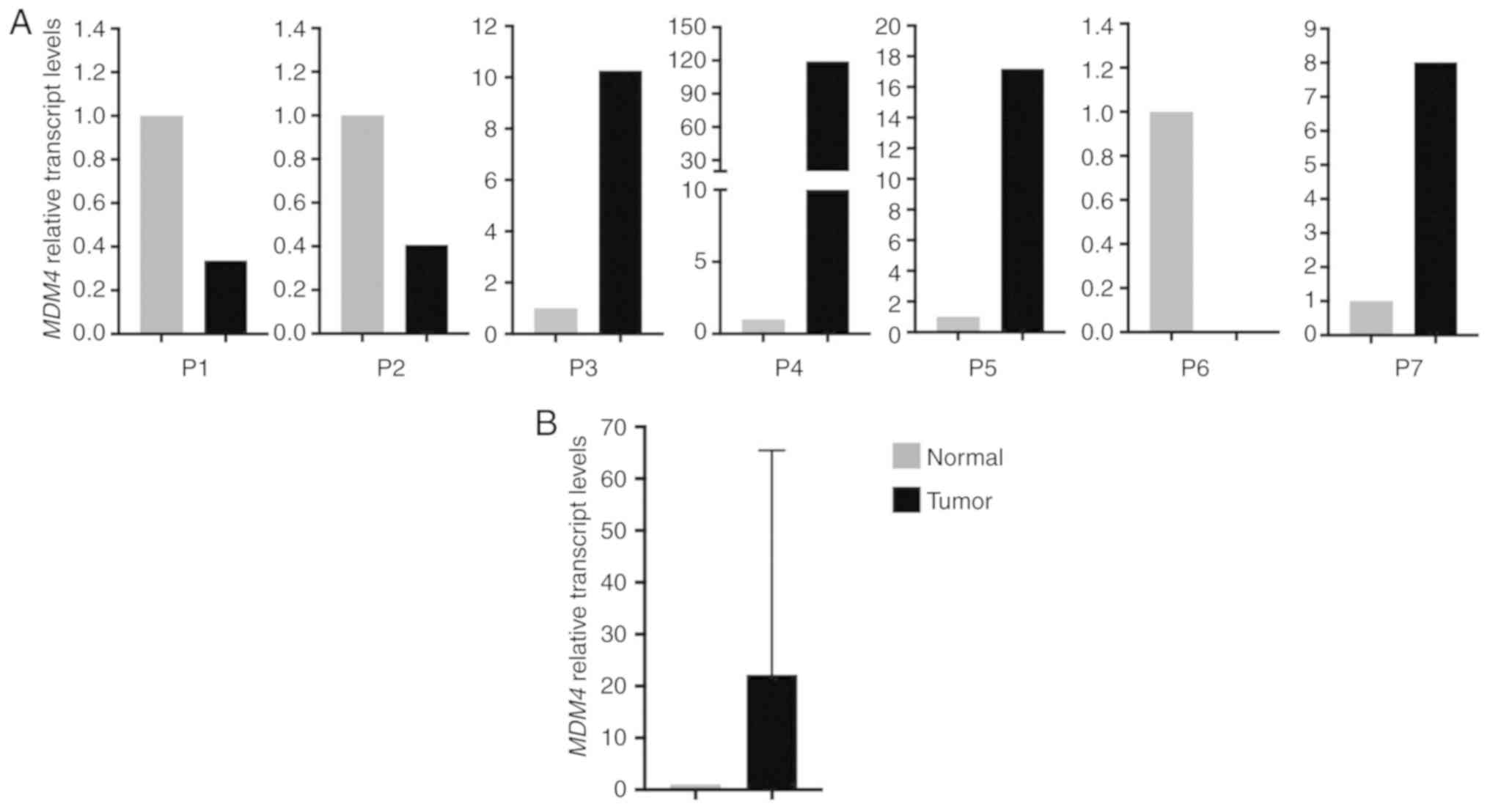
MDM4 transcipt levels are increased in
the majority of FL-HCC tumors
Previous studies have reported that MDM4 inhibits
the tumor suppressor activity of p53 by binding to its N-terminus
(22,23) and increased expression of MDM4 has
been reported in several other cancer types (25,28,34,35,43–45).
The present quantitative RT-PCR results indicated that the relative
transcript levels of MDM4 were increased in 4/7 tumors
(Fig. 4A). However, when all samples
were analyzed together, a statistically significant increase in
MDM4 transcript levels in FL-HCC tumors compared to their
non-neoplastic liver tissue (Mann-Whitney test, P=0.69, Fig. 4B) was not observed. This finding
indicated that MDM4 gene expression is increased in a
proportion of FL-HCC tumors.
MDM4 nuclear localization is
significantly increased in FL-HCC
Given the increased MDM4 transcript levels in
the majority of tumor samples, MDM4 protein expression was
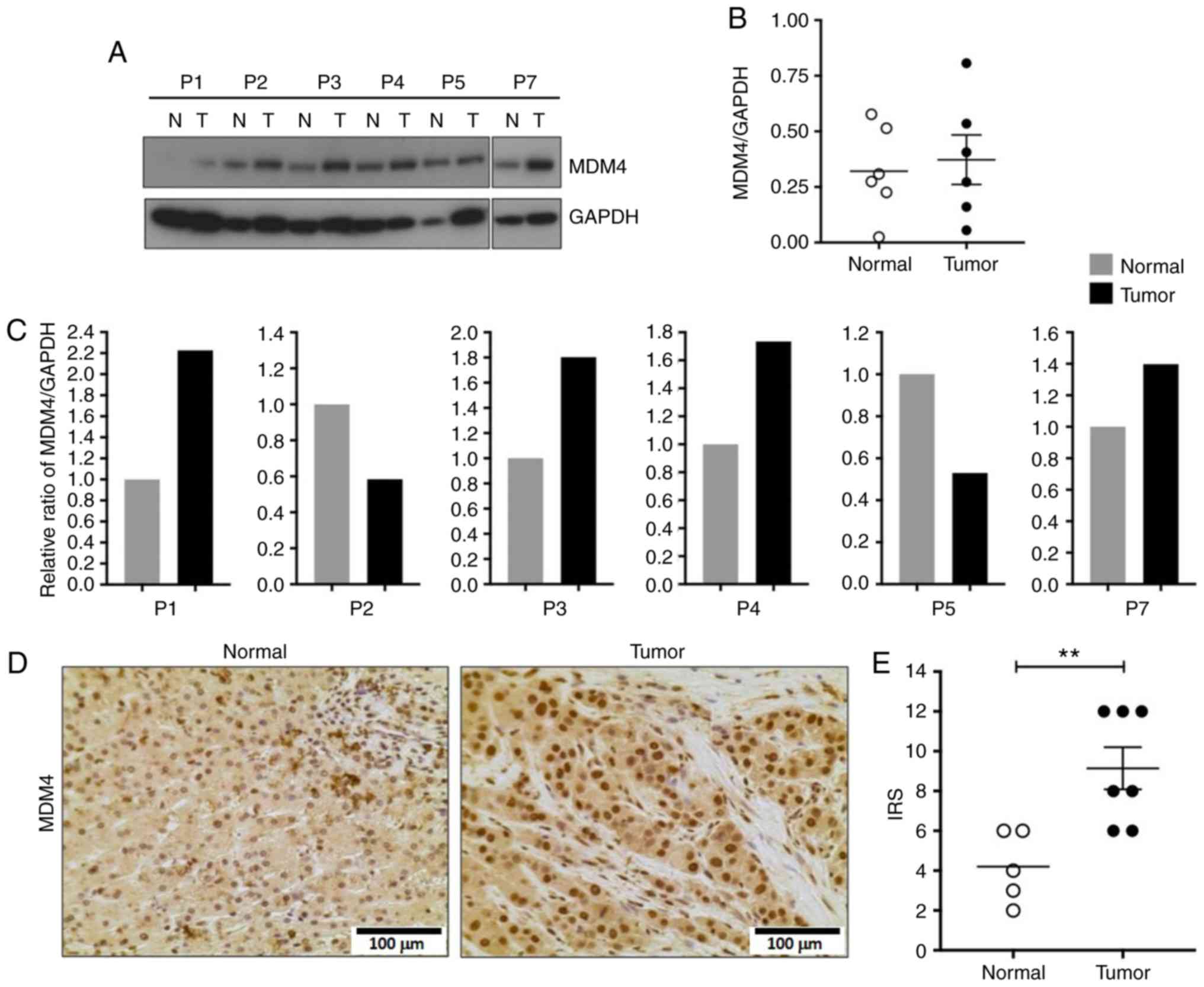
assessed. Western blot analysis revealed an increase in MDM4
protein levels in 4/6 tumor samples compared to the non-neoplastic
liver (Fig. 5A and C) but it did not
reach statistical significance when the samples were analyzed
together (Mann-Whitney test, P=0.93, Fig.
5B). However, there was a significant increase in nuclear MDM4
in tumor cells compared to non-neoplastic cells as revealed by IHC
(Mann-Whitney test, P=0.009; Fig. 5D and
E).
Discussion
In the present study, the DDR pathway components
p53, ATM, and MDM4 were examined in FL-HCC. The present findings
indicated that the DDR pathway may be activated given the increase
in nuclear p-ATM in FL-HCC. p53, the downstream target of ATM, was
also increased in tumor tissues without any difference in its
subcellular localization as assessed by IHC. In addition, the
activated or phosphorylated form of p53 (p-p53) did not localize to
the nucleus as predicted; instead p-p53 was localized to the
cytoplasmic compartment in tumor samples. Moreover, the transcript
levels of MDM4, a negative regulator of p53, were increased in 4/7
tumors, however, protein nuclear localization was increased in all
tumor samples (Table II).
| Table II.Summary of tumor IHC and MDM4
transcript levels. |
Table II.
Summary of tumor IHC and MDM4
transcript levels.
| Patient | DNAJB1-PRKACA
fusion present | MDM4 transcript
levels | MDM4 | Nuclear p-ATM | Cytoplasmic
p-ATM | Nuclear p-p53 | Cytoplasmic
p-p53 |
|---|
| P1 | Yes | 0.34 | +++ | + | ++ | − | +++ |
| P2 | Yes | 0.41 | ++ | − | ++ | − | − |
| P3 | Yes | 10.26 | +++ | + | ++ | + | ++ |
| P4 | No | 119.25 | +++ | ++ | ++ | ++ | ++ |
| P5 | Yes | 17.18 | ++ | ++ | + | − | + |
| P6 | Yes | 0.00006 | ++ | − | ++ | − | − |
| P7 | Yes | 8.01 | ++ | ++ | ++ | − | + |
FL-HCC was specifically focused on for the following
reasons: i) FL-HCC does not commonly arise in a background of liver
disease; ii) FL-HCC has a distinct histopathology compared to
conventional HCC; and iii) unlike many conventional HCC types,
FL-HCC does not contain multiple gene mutations, thereby
representing a potentially homogeneous tumor type. A recent advance
has been the revelation of a 400-kilobase pair deletion in
chromosome 19 resulting in DNJAB1-PRKACA fusion transcript and
protein in the majority of FL-HCC tumors (40,41). A
recent study using a mouse model demonstrated that expression of
the fusion protein in the liver is sufficient to result in FL-HCC,
thus demonstrating its role as an oncogenic driver (46). In the present study, the presence of
this fusion protein was confirmed in 6/7 of the tumor samples
examined. Given that our cohort only had a single tumor that did
not express the fusion protein, a conclusive determination was not
feasible without a larger sample size. This highlights the major
limitations of conducting cancer research on rare tumors such as
FL-HCC which include: i) Small sample size; ii) limited amount of
patient-derived tumor tissue; and iii) lack of in vitro or
in vivo models.
p53 is one of the key components of the DDR pathway
and its transcriptional activity has been revealed to prevent
aberrant oncogenic cell growth. Studies have reported that p53 is
mutated in ~50% of lung, liver, breast, brain, and prostate cancers
(24,47–49).
However, mutations in p53 have not been commonly observed in FL-HCC
(40,50). Upon detection of cellular stress and
DNA damage, ATM is activated which subsequently activates p53 to
induce downstream target genes to repair DNA damage, halt cell
growth, or induce apoptosis (51–54). In
fact, in the FL-HCC tumor tissue there was increased nuclear
localization of phosphorylated ATM (Fig.
3), indicating that the DNA damage repair process is activated
in these tumors. Under normal conditions, activated p53 or
phosphorylated p53 localizes to the nucleus where it performs its
transcriptional and tumor suppressor activity. However, despite the
increased phosphorylated ATM in the nucleus, the tumor samples
assessed in the present study lacked nuclear p-p53 as demonstrated
in Fig. 3A-C. In fact, phosphorylated
p53 preferentially localized to the cytoplasm of FL-HCC tumor cells
as opposed to the nucleus. These findings confirmed previously
published studies, which demonstrated a loss of tumor suppressive
function of non-mutant p53 via translocation to the cytoplasm in
several cancers (55–57). Notably, cancers lacking nuclear p53
are not responsive to chemotherapy or radiotherapy similar to
FL-HCC (55,56,58,59).
In the present study, the tumors demonstrated
activation of ATM which is an upstream regulator of p53. ATM
activation may be due to i) tumor cell DNA instability and attempt
at DNA repair or apoptosis or ii) a result of the toxic effects of
neo-adjuvant chemotherapy (60). In
either case, the expected response of p53 to elicit DNA damage
repair or apoptosis was not observed. Clinically, it is clear that
FL-HCC tumors are not responsive to conventional chemotherapy which
was also evident in our cohort given the high rate of tumor
progression and recurrence. On the other hand, assuming that ATM
activation is a result of the cell's recognition of DNA instability
following malignant transformation, the normal function of p53 is
still compromised. In our tumor cohort a significant nuclear
increase in the p53 negative regulator, MDM4, was observed.
Therefore, it is possible that increased MDM4 results in inhibition
of the normal function of p53 in these tumors. The exact mechanism
for increased MDM4 expression and nuclear localization is unclear,
although potential mechanisms may include increased gene
expression; decreased protein degradation due to changes in
post-transcriptional regulation; or alteration in
post-translational regulation of MDM4. For example, recent studies
have demonstrated that levels of miRNA-128 and miRNA-370 are
decreased in pancreatic and colon cancer, respectively (61,62). These
studies demonstrated that the aforementioned miRNAs can directly
inhibit MDM4 and promote cancer cell apoptosis. The role of miRNAs
in the regulation of MDM4 in FL-HCC is unknown at this time and
requires further research by examining the differential expression
of known MDM4-specific miRNA in tumor cells compared to normal
hepatocytes.
In the cancers which do not have mutated p53, there
is evidence that tumor cells inhibit the normal function of p53 and
prevent the cell from committing to DNA repair or apoptosis. Even
in tumors with p53 mutation, studies have focused on
re-establishing the normal function of p53 despite the presence of
its mutated form. For example, in vitro studies have
revealed that small molecules can reverse the aberrant function of
mutant p53 and subsequently induce apoptosis (63–65).
Therefore, the idea of reactivating or restoring the function of
p53 for the treatment of cancer has been gaining interest (12,66). One
strategy to restore the normal function of non-mutated p53 is to
target its negative regulators such as MDM4. MDM4 induces its
inhibitory effect by binding to the N-terminus transactivation
domain of p53 and affecting its transcriptional activity (22,23,67).
Recent studies have revealed that MDM4 is frequently overexpressed
in a number of cancer types lacking p53 mutations including breast,
melanoma, retinoblastoma, acute myeloid leukemia, and esophageal
squamous cell carcinoma (25,28,43–45,68,69).
Therefore, in these cancers, inhibition of MDM4 activity has been
an appealing therapeutic strategy to reactivate the function of p53
and ultimately control aberrant cell proliferation and tumor
progression. In light of these findings, there is an interest in
identifying small molecules or peptides that specifically target
MDM4. In fact, MDM4 inhibitors have exhibited promising efficacy in
both in vitro and in vivo models of cancers. Wang
et al demonstrated that the MDM4 inhibitor, NSC207895,
restored normal function of p53 in breast cancer cell lines and
triggered cell apoptosis (70).
Likewise, knockdown of MDM4 in breast cancer cell lines and
xenograft models was revealed to induce senescence of cancer cells
(29). In addition, in retinoblastoma
cells, MDM4 inhibitor, SJ-172550, effectively triggered cell death
(71). Lastly, in melanoma cells,
known to be resistant to BRAF (B-Raf proto oncogene, serine
threonine kinase) inhibitors, the inhibition of p53-MDM4
interaction using the MDM4 inhibitor, SAH-p53-8, exhibited
effective activation of p53 and reduction in cell growth (28). Collectively, these studies indicated
that inhibiting MDM4 can restore p53 activity in cancers lacking
p53 mutations, thereby providing a promising new therapeutic
strategy. To the best of our knowledge, the present study is the
first to specifically examine MDM4 in FL-HCC. Whether p53
dysregulation is directly due to MDM4 activity in this tumor type
is yet unclear, however, given the existing evidence in other
cancers, it is a plausible hypothesis. In conclusion, our finding
of increased nuclear MDM4 in FL-HCC tumors unveils a potential new
target for the development of novel treatments for this highly
lethal cancer.
Supplementary Material
Supporting Data
Acknowledgements
We would like to thank Dr Amanda Dios, Kaly Mueller,
Eric Granucci, Dr Ina Kycia, Dr Sanjeev Vasudevan and Dr Sarah
Woodfield for their scientific advice on this study.
Funding
The present research was funded by CHMC Surgical
Foundation, Inc.
Author contributions
AK and KV designed and AK performed all experiments.
JP and ARPA reviewed the pathology slides and quantified the
staining sections. AK, MJL, SSK, GSV and KV analyzed all data. AK
and KV wrote the manuscript and all co-authors reviewed the final
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Ethics approval and consent to
participate
The use of human tissue in the present study was
approved by the Institutional Review Board at Boston Children's
Hospital. Patients and/or patient's guardians provided consent for
the use of tissue in the present study.
Patient consent for publication
Not applicable.
Competing interests
The authors do not have any personal, professional,
or financial relationships that would constitute a competing
interest in regards to this work.
References
|
1
|
Schlaeger C, Longerich T, Schiller C,
Bewerunge P, Mehrabi A, Toedt G, Kleeff J, Ehemann V, Eils R,
Lichter P, et al: Etiology-dependent molecular mechanisms in human
hepatocarcinogenesis. Hepatology. 47:511–520. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB and Davila JA: Is
fibrolamellar carcinoma different from hepatocellular carcinoma? A
US population-based study. Hepatology. 39:798–803. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mavros MN, Mayo SC, Hyder O and Pawlik TM:
A systematic review: Treatment and prognosis of patients with
fibrolamellar hepatocellular carcinoma. J Am Coll Surg.
215:820–830. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kakar S, Chen X, Ho C, Burgart LJ, Sahai
V, Dachrut S, Yabes A, Jain D and Ferrell LD: Chromosomal changes
in fibrolamellar hepatocellular carcinoma detected by array
comparative genomic hybridization. Mod Pathol. 22:134–141. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Craig JR, Peters RL, Edmondson HA and
Omata M: Fibrolamellar carcinoma of the liver: A tumor of
adolescents and young adults with distinctive clinico-pathologic
features. Cancer. 46:372–379. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kakar S, Burgart LJ, Batts KP, Garcia J,
Jain D and Ferrell LD: Clinicopathologic features and survival in
fibrolamellar carcinoma: Comparison with conventional
hepatocellular carcinoma with and without cirrhosis. Mod Pathol.
18:1417–1423. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Edmondson HA: Differential diagnosis of
tumors and tumor-like lesions of liver in infancy and childhood.
AMA J Dis Child. 91:168–186. 1956.PubMed/NCBI
|
|
8
|
Sanyal AJ, Yoon SK and Lencioni R: The
etiology of hepatocellular carcinoma and consequences for
treatment. Oncologist. 15 (Suppl 4):S14–S22. 2010. View Article : Google Scholar
|
|
9
|
Lafaro KJ and Pawlik TM: Fibrolamellar
hepatocellular carcinoma: Current clinical perspectives. J
Hepatocell Carcinoma. 2:151–157. 2015.PubMed/NCBI
|
|
10
|
Lane DP: Cancer. p53, guardian of the
genome. Nature. 358:15–16. 1992. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Efeyan A and Serrano M: p53: Guardian of
the genome and policeman of the oncogenes. Cell Cycle. 6:1006–1010.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Selivanova G: Wild type p53 reactivation:
From lab bench to clinic. FEBS Lett. 588:2628–2638. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shiloh Y: ATM: Ready, set, go. Cell Cycle.
2:116–117. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shiloh Y: ATM and related protein kinases:
Safeguarding genome integrity. Nat Rev Cancer. 3:155–168. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Banin S, Moyal L, Shieh S, Taya Y,
Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y
and Ziv Y: Enhanced phosphorylation of p53 by ATM in response to
DNA damage. Science. 281:1674–1677. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Polyak K, Xia Y, Zweier JL, Kinzler KW and
Vogelstein B: A model for p53-induced apoptosis. Nature.
389:300–305. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livingstone LR, White A, Sprouse J,
Livanos E, Jacks T and Tlsty TD: Altered cell cycle arrest and gene
amplification potential accompany loss of wild-type p53. Cell.
70:923–935. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sengupta S and Harris CC: p53: Traffic cop
at the crossroads of DNA repair and recombination. Nat Rev Mol Cell
Biol. 6:44–55. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Marine JC: MDM2 and MDMX in cancer and
development. Curr Top Dev Biol. 94:45–75. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marine JC and Jochemsen AG: Mdmx as an
essential regulator of p53 activity. Biochem Biophys Res Commun.
331:750–760. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Haupt Y, Maya R, Kazaz A and Oren M: Mdm2
promotes the rapid degradation of p53. Nature. 387:296–299. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shvarts A, Bazuine M, Dekker P, Ramos YF,
Steegenga WT, Merckx G, van Ham RC, van der Houven van Oordt W, van
der Eb AJ and Jochemsen AG: Isolation and identification of the
human homolog of a new p53-binding protein, Mdmx. Genomics.
43:34–42. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shvarts A, Steegenga WT, Riteco N, van
Laar T, Dekker P, Bazuine M, van Ham RC, van der Houven van Oordt
W, Hateboer G, van der Eb AJ and Jochemsen AG: MDMX: A novel
p53-binding protein with some functional properties of MDM2. EMBO
J. 15:5349–5357. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Leroy B, Anderson M and Soussi T: TP53
mutations in human cancer: Database reassessment and prospects for
the next decade. Hum Mutat. 35:672–688. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li L, Tan Y, Chen X, Xu Z, Yang S, Ren F,
Guo H, Wang X, Chen Y, Li G and Wang H: MDM4 overexpressed in acute
myeloid leukemia patients with complex karyotype and wild-type
TP53. PLoS One. 9:e1130882014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Quintás-Cardama A, Hu C, Qutub A, Qiu YH,
Zhang X, Post SM, Zhang N, Coombes K and Kornblau SM: p53 pathway
dysfunction is highly prevalent in acute myeloid leukemia
independent of TP53 mutational status. Leukemia. 31:1296–1305.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li Q and Lozano G: Molecular pathways:
Targeting Mdm2 and Mdm4 in cancer therapy. Clin Cancer Res.
19:34–41. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gembarska A, Luciani F, Fedele C, Russell
EA, Dewaele M, Villar S, Zwolinska A, Haupt S, de Lange J, Yip D,
et al: MDM4 is a key therapeutic target in cutaneous melanoma. Nat
Med. 18:1239–1247. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Haupt S, Buckley D, Pang JM, Panimaya J,
Paul PJ, Gamell C, Takano EA, Lee YY, Hiddingh S, Rogers TM, et al:
Targeting Mdmx to treat breast cancers with wild-type p53. Cell
Death Dis. 6:e18212015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kan Z, Zheng H, Liu X, Li S, Barber TD,
Gong Z, Gao H, Hao K, Willard MD, Xu J, et al: Whole-genome
sequencing identifies recurrent mutations in hepatocellular
carcinoma. Genome Res. 23:1422–1433. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kunst C, Haderer M, Heckel S, Schlosser S
and Müller M: The p53 family in hepatocellular carcinoma. Transl
Cancer Res. 5:632–638. 2016. View Article : Google Scholar
|
|
32
|
Ward SC and Waxman S: Fibrolamellar
carcinoma: A review with focus on genetics and comparison to other
malignant primary liver tumors. Semin Liver Dis. 31:61–70. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sorenson EC, Khanin R, Bamboat ZM, Cavnar
MJ, Kim TS, Sadot E, Zeng S, Greer JB, Seifert AM, Cohen NA, et al:
Genome and transcriptome profiling of fibrolamellar hepatocellular
carcinoma demonstrates p53 and IGF2BP1 dysregulation. PLoS One.
12:e01765622017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cancer Genome Atlas Research Network.
Electronic address, . wheeler@bcm.edu; Cancer Genome AtlasResearch
Network: Comprehensive and integrative genomic characterization of
hepatocellular carcinoma. Cell. 169:1327–1341.e23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Arai Y, Honda S, Haruta M, Kasai F,
Fujiwara Y, Ohshima J, Sasaki F, Nakagawara A, Horie H, Yamaoka H,
et al: Genome-wide analysis of allelic imbalances reveals 4q
deletions as a poor prognostic factor and MDM4 amplification at
1q32.1 in hepatoblastoma. Genes Chromosomes Cancer. 49:596–609.
2010.PubMed/NCBI
|
|
36
|
LaQuaglia MJ, Grijalva JL, Mueller KA,
Perez-Atayde AR, Kim HB, Sadri-Vakili G and Vakili K: YAP
subcellular localization and Hippo pathway transcriptome analysis
in pediatric hepatocellular carcinoma. Sci Rep. 6:302382016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fedchenko N and Reifenrath J: Different
approaches for interpretation and reporting of immunohistochemistry
analysis results in the bone tissue-a review. Diagn Pathol.
9:2212014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hemming AW, Langer B, Sheiner P, Greig PD
and Taylor BR: Aggressive surgical management of fibrolamellar
hepatocellular carcinoma. J Gastrointest Surg. 1:342–346. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stevens WR, Johnson CD, Stephens DH and
Nagorney DM: Fibrolamellar hepatocellular carcinoma: Stage at
presentation and results of aggressive surgical management. AJR Am
J Roentgenol. 164:1153–1158. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Honeyman JN, Simon EP, Robine N,
Chiaroni-Clarke R, Darcy DG, LimI I, Gleason CE, Murphy JM,
Rosenberg BR, Teegan L, et al: Detection of a recurrent
DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular
carcinoma. Science. 343:1010–1014. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Graham RP, Jin L, Knutson DL, Kloft-Nelson
SM, Greipp PT, Waldburger N, Roessler S, Longerich T, Roberts LR,
Oliveira AM, et al: DNAJB1-PRKACA is specific for fibrolamellar
carcinoma. Mod Pathol. 28:822–829. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Turenne GA, Paul P, Laflair L and Price
BD: Activation of p53 transcriptional activity requires ATM's
kinase domain and multiple N-terminal serine residues of p53.
Oncogene. 20:5100–5110. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Haupt S, Vijayakumaran R, Miranda PJ,
Burgess A, Lim E and Haupt Y: The role of MDM2 and MDM4 in breast
cancer development and prevention. J Mol Cell Biol. 9:53–61. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang H, Hu L, Qiu W, Deng T, Zhang Y,
Bergholz J and Xiao ZX: MDMX exerts its oncogenic activity via
suppression of retinoblastoma protein. Oncogene. 34:5560–5569.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Carneiro A, Isinger A, Karlsson A,
Johansson J, Jönsson G, Bendahl PO, Falkenback D, Halvarsson B and
Nilbert M: Prognostic impact of array-based genomic profiles in
esophageal squamous cell cancer. BMC Cancer. 8:982008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tomasini MD, Wang Y, Karamafrooz A, Li G,
Beuming T, Gao J, Taylor SS, Veglia G and Simon SM: Conformational
LANDSCAPE of the PRKACA-DNAJB1 chimeric kinase, the driver for
fibrolamellar hepatocellular carcinoma. Sci Rep. 8:7202018.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Petitjean A, Mathe E, Kato S, Ishioka C,
Tavtigian SV, Hainaut P and Olivier M: Impact of mutant p53
functional properties on TP53 mutation patterns and tumor
phenotype: Lessons from recent developments in the IARC TP53
database. Hum Mutat. 28:622–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Olivier M, Hollstein M and Hainaut P: TP53
mutations in human cancers: Origins, consequences, and clinical
use. Cold Spring Harb Perspect Biol. 2:a0010082010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lawrence MS, Stojanov P, Polak P, Kryukov
GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH,
Roberts SA, et al: Mutational heterogeneity in cancer and the
search for new cancer-associated genes. Nature. 499:214–218. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xu L, Hazard FK, Zmoos AF, Jahchan N,
Chaib H, Garfin PM, Rangaswami A, Snyder MP and Sage J: Genomic
analysis of fibrolamellar hepatocellular carcinoma. Hum Mol Genet.
24:50–63. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Finlay CA, Hinds PW and Levine AJ: The p53
proto-oncogene can act as a suppressor of transformation. Cell.
57:1083–1093. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Harper JW, Adami GR, Wei N, Keyomarsi K
and Elledge SJ: The p21 Cdk-interacting protein Cip1 is a potent
inhibitor of G1 cyclin-dependent kinases. Cell. 75:805–816. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kastenhuber ER and Lowe SW: Putting p53 in
context. Cell. 170:1062–1078. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mills KD: Tumor suppression: Putting p53
in context. Cell Cycle. 12:3461–3462. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
O'Brate A and Giannakakou P: The
importance of p53 location: Nuclear or cytoplasmic zip code? Drug
Resist Updat. 6:313–322. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sembritzki O, Hagel C, Lamszus K, Deppert
W and Bohn W: Cytoplasmic localization of wild-type p53 in
glioblastomas correlates with expression of vimentin and glial
fibrillary acidic protein. Neuro Oncol. 4:171–178. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Green DR and Kroemer G: Cytoplasmic
functions of the tumour suppressor p53. Nature. 458:1127–1130.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Moll UM, Riou G and Levine AJ: Two
distinct mechanisms alter p53 in breast cancer: Mutation and
nuclear exclusion. Proc Natl Acad Sci USA. 89:7262–7266. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Bosari S, Viale G, Roncalli M, Graziani D,
Borsani G, Lee AK and Coggi G: p53 gene mutations, p53 protein
accumulation and compartmentalization in colorectal adenocarcinoma.
Am J Pathol. 147:790–798. 1995.PubMed/NCBI
|
|
60
|
Vici P, Di Benedetto A, Ercolani C,
Pizzuti L, Di Lauro L, Sergi D, Sperati F, Terrenato I, Dattilo R,
Botti C, et al: Predictive significance of DNA damage and repair
biomarkers in triple-negative breast cancer patients treated with
neoadjuvant chemotherapy: An exploratory analysis. Oncotarget.
6:42773–42780. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Shen X, Zuo X, Zhang W, Bai Y, Qin X and
Hou N: MiR-370 promotes apoptosis in colon cancer by directly
targeting MDM4. Oncol Lett. 15:1673–1679. 2018.PubMed/NCBI
|
|
62
|
Han H, Wang L, Xu J and Wang A: miR-128
induces pancreas cancer cell apoptosis by targeting MDM4. Exp Ther
Med. 15:5017–5022. 2018.PubMed/NCBI
|
|
63
|
Zache N, Lambert JM, Rökaeus N, Shen J,
Hainaut P, Bergman J, Wiman KG and Bykov VJ: Mutant p53 targeting
by the low molecular weight compound STIMA-1. Mol Oncol. 2:70–80.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Bykov VJ, Issaeva N, Shilov A, Hultcrantz
M, Pugacheva E, Chumakov P, Bergman J, Wiman KG and Selivanova G:
Restoration of the tumor suppressor function to mutant p53 by a
low-molecular-weight compound. Nat Med. 8:282–288. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hiraki M, Hwang SY, Cao S, Ramadhar TR,
Byun S, Yoon KW, Lee JH, Chu K, Gurkar AU, Kolev V, et al:
Small-molecule reactivation of mutant p53 to wild-type-like p53
through the p53-Hsp40 regulatory axis. Chem Biol. 22:1206–1216.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Bossi G and Sacchi A: Restoration of
wild-type p53 function in human cancer: Relevance for tumor
therapy. Head Neck. 29:272–284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Stad R, Little NA, Xirodimas DP, Frenk R,
van der Eb AJ, Lane DP, Saville MK and Jochemsen AG: Mdmx
stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Rep.
2:1029–1034. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Garcia D, Warr MR, Martins CP, Brown
Swigart L, Passegué E and Evan GI: Validation of MdmX as a
therapeutic target for reactivating p53 in tumors. Genes Dev.
25:1746–1757. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Danovi D, Meulmeester E, Pasini D,
Migliorini D, Capra M, Frenk R, de Graaf P, Francoz S, Gasparini P,
Gobbi A, et al: Amplification of Mdmx (or Mdm4) directly
contributes to tumor formation by inhibiting p53 tumor suppressor
activity. Mol Cell Biol. 24:5835–5843. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wang H, Ma X, Ren S, Buolamwini JK and Yan
C: A small-molecule inhibitor of MDMX activates p53 and induces
apoptosis. Mol Cancer Ther. 10:69–79. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Reed D, Shen Y, Shelat AA, Arnold LA,
Ferreira AM, Zhu F, Mills N, Smithson DC, Regni CA, Bashford D, et
al: Identification and characterization of the first small molecule
inhibitor of MDMX. J Biol Chem. 285:10786–10796. 2010. View Article : Google Scholar : PubMed/NCBI
|