Introduction
Cholangiocarcinoma (CCA) originates from
intrahepatic or extrahepatic bile duct epithelial cells and is
classified into intrahepatic CCA (iCCA), perihilar CCA (pCCA), and
distal CCA (dCCA) according to the anatomic location (1,2). Among the
majority of CCA tumors, pCCA accounts for 60–70%, dCCA accounts for
20–30% and iCCA accounts for 5–10%. CCA is recognized as the second
most commonly diagnosed primary liver tumor and accounts for
approximately 1–15% of all hepatobiliary malignancies (3). Although the average incidence of CCA is
low, early diagnosis and treatment of CCA are difficult, and the
overall patient prognosis is poor (4). Recently, iCCA has become the leading
cause of death related to primary liver tumor (5). Systemic drug therapy is currently
limited for patients with advanced or metastatic CCA, while
surgical treatment is suitable only for those with early-stage CCA,
which has a high risk of recurrence (6,7). The
median survival time of patients with advanced CCA is less than 2
years, and the 5-year survival rate is only 10% (3,8). Searching
for genetic drivers that affect the occurrence and progression of
CCA is important for exploring the molecular diagnosis and targeted
therapy (1). In recent years,
biomarker research has achieved progress in the prediction,
treatment and prognosis of CCA. For example, KRAS mutations and
PRKACB fusion genes have been identified in pCCA and dCCA, and
somatic mutations of isocitrate dehydrogenase (IDH) have been
identified in iCCA (9). In addition,
inducible nitric oxide synthase (iNOS) has been involved in the
occurrence of CCA through an inflammation-dependent manner
(10). However, due to strong genetic
heterogeneity, the current understanding of the molecular
mechanisms of CCA is still not comprehensive. In particular,
understanding of the genetic variations that promote CCA initiation
and development are still fragmented. Moreover, the key driver
genes of carcinogenesis remain unknown (4,11).
Therefore, studying the pathogenesis of CCA and identifying hub
genes that are involved in the development of CCA remain a major
challenge.
The Cancer Genome Atlas (TCGA) is a publicly
sponsored project with the purpose of classifying and identifying
major carcinogenic genomic alterations among large cohorts of more
than 30 human tumors. To perform an integrated analysis of cancer
genome profiles, high-throughput technologies relying on the use of
microarrays and next-generation sequencing methods were applied in
TCGA (12). RNA sequencing (RNAseq)
has become useful for transcriptome (total RNA) profiling and
obtaining accurate strand information. RNAseq is a method that is
conductive to the application of a systematic comprehensive study
of differentially expressed gene interactions and related signaling
pathways with high precision. Moreover, protein-protein interaction
(PPI) networks are useful for distinguishing hub genes, which are
defined as genes with a high degree of connectivity that play an
essential role in stabilizing the PPI network structure (13,14). There
are numerous oncology studies based on TCGA. From the perspective
of CCA, Wang et al thoroughly studied the lncRNA-miRNA-mRNA
ceRNA network and identified three lncRNAs, COL18A1-AS1, SLC6A1-AS1
and HULC, as being significantly associated with overall CCA
patient survival (15). However, in
the present study, we focused on identifying hub genes within the
PPI and exploring their potential roles in CCA on the basis of TCGA
combined with multiple datasets.
In the present research, transcriptomic iCCA data
from TCGA were utilized to identify differentially expressed
protein-coding genes (DEGs) between iCCA and normal tissues. Then,
we executed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and
Genomes (KEGG) enrichment analysis to study alterations in
biological functions and signaling pathways of iCCA. PPI network
construction was performed, followed by identification of hub
genes. Moreover, we identified the differential expression of hub
genes by analyzing transcriptomic CCA data from several open-access
databases, including Gene Expression Omnibus (GEO) database and
ArrayExpress Archive of Functional Genomics Data (ArrayExpress).
Further, we performed quantitative polymerase chain reaction (qPCR)
in the laboratory to verify these hub genes. Finally, we executed
survival analysis of the identified hub genes. The objective of
this study was to understand CCA carcinogenesis by exploring the
genetic changes involved in disease progression and to identify
potential biomarkers that may be helpful for predicting the
prognosis of iCCA patients.
Materials and methods
Acquisition of transcriptome data and
identification of DEGs
Data for CCA mRNA expression were downloaded from
TCGA database (https://portal.gdc.cancer.gov/, RNA-seq, Illumina) on
July 6, 2018. Practical Extraction and Reporting Language (Perl)
was utilized for sample information extraction, mRNA expression
matrix generation, and gene symbol annotation. Only samples of
primary site of liver and intrahepatic bile ducts were included for
subsequent analysis, Statistical softwareR (version 3.4.4) and the
‘DEseq’ package from Bioconductor were used to perform significance
analysis of the DEGs between CCA samples and adjacent noncancerous
tissues (16,17). Genes with an absolute value of log2
fold change (log2FC) >2 and a corrected P-value <0.0001 were
defined as DEGs.
Functional and pathway enrichment
analyses
GO term enrichment analysis was applied to analyze
the biological significance of DEGs, which includes biological
processes (BP), cellular components (CC) and molecular functions
(MF), based on the GO online platform David (https://david.ncifcrf.gov/, date of access: 2019/5/7,
species: Human). GO visualization was achieved by the R package
‘GOplot’ (18). KEGG pathway
enrichment analysis was applied based on the online platform David.
Critical pathways enriched in DEGs were identified. Visualization
of KEGG results was conducted by R package ‘ggplot2’. P<0.05 was
considered statistically significant for both GO and KEGG
analysis.
PPI network analysis
Proteins usually perform biological functions
synergistically. Strong relationships have been shown to exist
between PPIs and the biological functions of gene/protein clusters
(19). Therefore, PPIs must be
explored by considering functional groups. PPI network analysis is
helpful for distinguishing hub genes among a cluster of DEGs that
are implicated in CCA progression based on their interaction
levels. PPI information of DEGs was obtained from the STRING
database; highest confidence of 0.900 was chosen (version 11.0,
https://string-db.org/, data of access:
2019/5/7). The PPI networks for upregulated genes was constructed
using Cytoscape3.6.1 software (https://cytoscape.org/). The top 15 genes with the
highest degree of connectivity were defined as hub genes.
Identification of hub genes
CCA-related transcriptomic datasets were obtained
from GEO (GSE76297 and GSE26566) and ArrayExpress (E-GEOD-32879 and
E-GEOD-45001) (20–24). R (version 3.4.4) and the ‘Limma’
package of Bioconductor were used for identification of the DEGs
(25). A P-value <0.05 was
considered statistically significant. Finally, hub genes in TCGA
were compared with the DEGs acquired from other 4 datasets. Hub
genes with similar differential expression among 5 datasets were
selected for further analysis. A Venn diagram indicating the
intersection of multiple datasets was made online (http://bioinformatics.psb.ugent.be/cgi-bin/liste/Venn/calculate_venn.htpl).
Statistical analysis
To examine the classification effect of hub genes on
cholangiocarcinoma and normal tissue, receiver operating
characteristic (ROC) curves and area under the curve (AUC) were
calculated by R package ‘pROC’ (26).
Furthermore, clinical data of iCCA were downloaded from TCGA. We
divided patients into two groups based on tumor stage: Stage I+II
and stage III+IV. The association between hub gene expression and
tumor stage was evaluated by Mann-Whitney U test. A P<0.05 was
considered statistically significant. Finally, ‘survival’ package
of Bioconductor was used to generate overall survival curves
(27,28). Perl was used to extract the lifetime
of each patient from the clinical cart downloaded from TCGA.
Clinical data of 118 patients with cholangiocarcinoma were
downloaded from PubMed Central (11).
Matching sample information was obtained from GEO dataset: GSE89749
(11). For each dataset, the patients
were divided into two groups using the median gene expression value
as the cut-off value. The relationship between patient overall
survival and expression level of hub genes was tested by Cox
proportional-hazards model. P<0.05 was considered statistically
significant.
RT-qPCR
Tissue samples were collected as pairs, i.e., tumor
tissue and adjacent normal tissue, from 10 patients with iCCA
undergoing surgery at Peking Union Medical College Hospital. A
total of 6 male and 4 female patients with mean age of 62 (range,
54–68) years were included. The collected tissue samples were
stored in a refrigerator at −80°C. All patients were enrolled from
November 2018 to April 2019. The study was approved by the Clinical
Research Ethics Committee of Peking Union Medical College Hospital.
Each patient provided a written informed signed consent. Total RNA
was isolated from each sample with Trizol LS reagent (Invitrogen;
Thermo Fisher Scientific, Inc.), and then used for cDNA synthesis
using oligo(dT)primers and SuperScript™ III Reverse Transcriptase
(Invitrogen; Thermo Fisher Scientific, Inc.). PCR Master Mix (2X)
(Superarray) and Applied Biosystems QuantStudio5 Real-time PCR
system (Thermo Fisher Scientific, Inc.) were utilized for RT-qPCR.
The sequences of primers for selected hub genes and housekeeping
gene (β-actin) are shown in Table
SI.
Visualization of differential
expression
For hub genes validated by qRT-PCR, R package
‘ggpubr’ (https://rpkgs.datanovia.com/ggpubr/index.html) was
used to visualize gene expression based on the expression profile
of DEGs in TCGA and the results of qRT-PCR. A t-test was used to
calculate differences between groups. P<0.05 was considered
statistically significant.
Results
Identification of DEGs in CCA and
normal tissues
The transcriptomic dataset of CCA and the
corresponding clinical cart were downloaded from the TCGA database.
Patients with lesion of primary site of liver and intrahepatic bile
ducts, i.e., patients with iCCA, were included for further
analysis. Therefore, a total of 33 cases were acquired, including
19 female and 14 male patients. Forty-one samples were acquired in
total, including 33 tumor tissue samples and 8 normal tissue
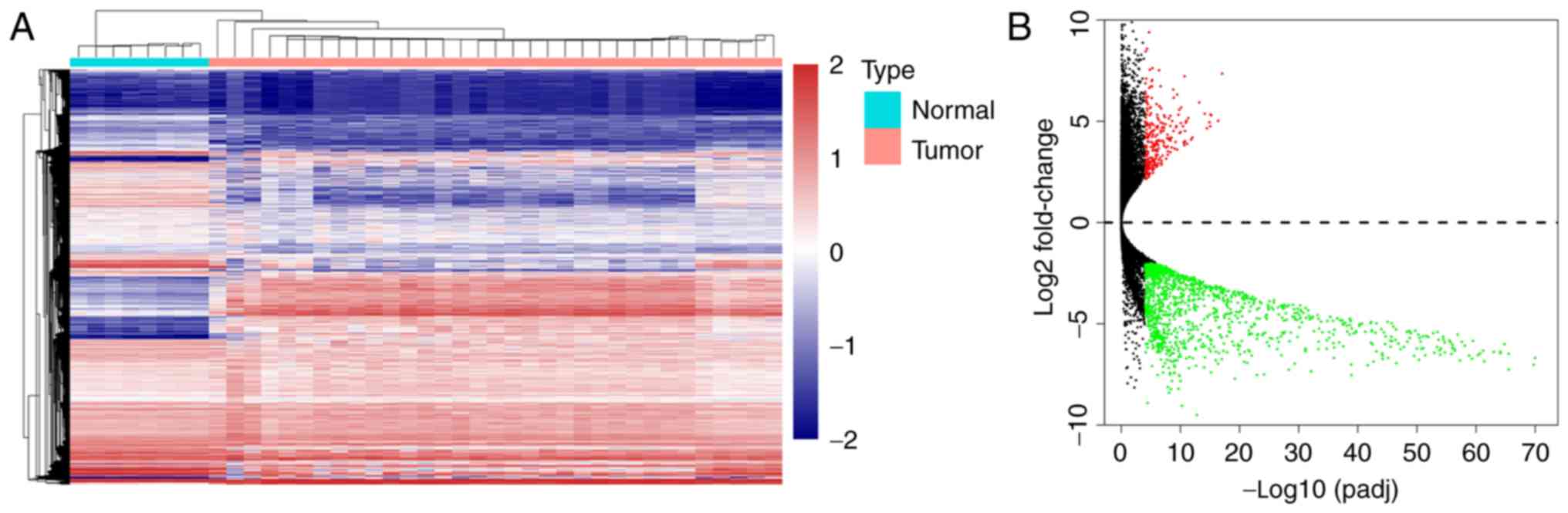
samples. A total of 1,463 DEGs (log2FC >2, corrected
P<0.0001) were acquired, including 267 significantly upregulated
DEGs and 1,196 significantly downregulated DEGs. A heatmap and a
volcano plot showing the expression levels of these genes are shown
in Fig. 1.
Functional and pathway enrichment
analyses of DEGs
GO and KEGG pathway enrichment analyses were
conducted to explore the functional characteristics of the DEGs.
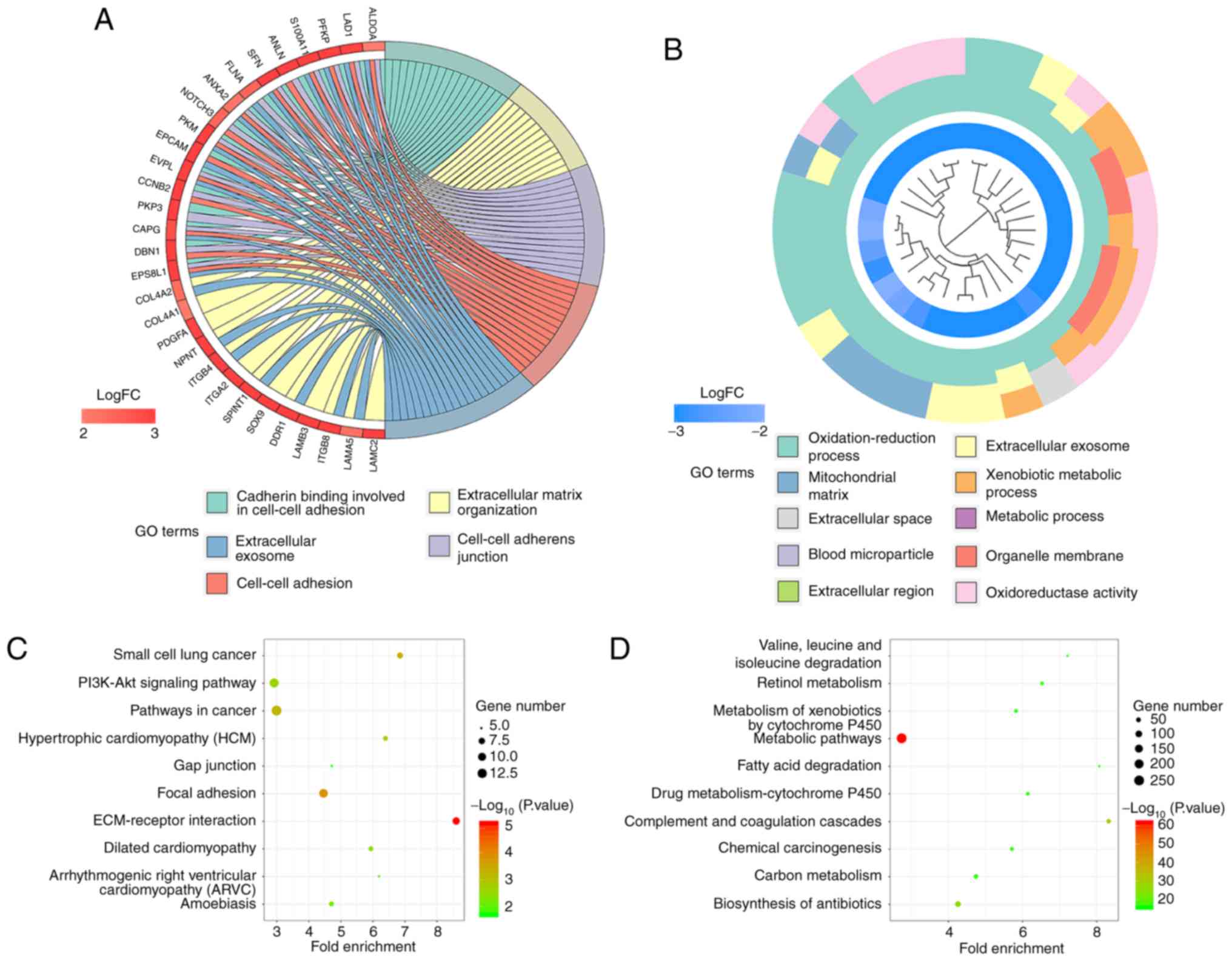
The GO analysis results revealed that the upregulated DEGs were
significantly enriched in ‘extracellular matrix organization’,
‘cell-cell adhesion’, ‘cell adhesion’, ‘epithelial cell
morphogenesis involved in placental branching and mitotic spindle
assembly’ in terms of BP. Regarding MF, the upregulated DEGs were
enriched in ‘cadherin binding involved in cell-cell adhesion’,
‘structural molecule activity’, ‘collagen binding’, ‘protein
binding’ and ‘signal transducer activity’. Under CC, the
upregulated DEGs were enriched in ‘cell-cell adherens junction’,
‘extracellular exosome’, ‘midbody, cell-cell junction’ and
‘cytoplasmic microtubule’. For the downregulated DEGs, significant
enrichment was observed in the ‘oxidation-reduction process’,
‘xenobiotic metabolic process’, ‘metabolic process’, ‘steroid
metabolic process’ and ‘platelet degranulation’ under BP. For MF,
the downregulated DEGs were significantly enriched in
‘oxidoreductase activity’, ‘monooxygenase activity’, ‘iron ion
binding’, ‘oxidoreductase activity acting on paired donors, with
the incorporation of or reduction in molecular oxygen’ and
‘electron carrier activity’. For CC, the DEGs were enriched in
‘extracellular exosome’, ‘blood microparticle’, ‘organelle
membrane’, ‘mitochondrial matrix’ and ‘extracellular region’. In
addition, the KEGG analysis results showed that the upregulated
DEGs were significantly enriched in ‘ECM-receptor interactions’,
‘focal adhesion’, ‘small cell lung cancer’, ‘pathways in cancer’
and ‘hypertrophic cardiomyopathy (HCM)’. Meanwhile, the
downregulated DEGs were enriched in ‘metabolic pathways’,
‘complement and coagulation cascades’, ‘biosynthesis of
antibiotics’, ‘retinol metabolism’ and ‘fatty acid degradation’.
The enriched GO terms and KEGG pathways are shown in Fig. 2 and Tables
SII–SV.
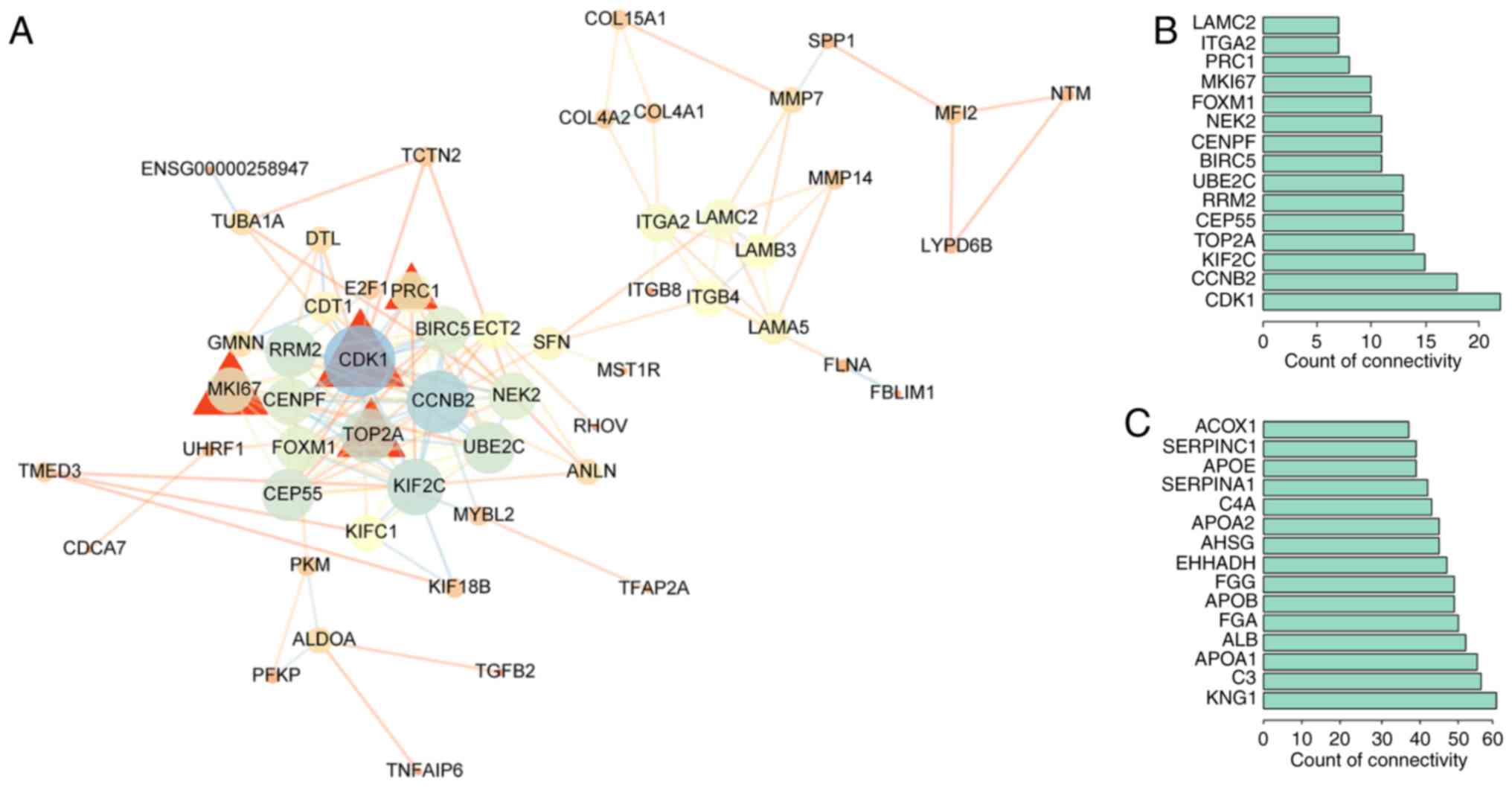
Construction of the PPI network and
hub gene identification
PPI network analysis can be used to distinguish
critical hub genes among a group of DEGs. Therefore, the STRING
database was used to conduct the PPI network analysis. PPI networks
for the upregulated genes were constructed by Cytoscape 3.6.1
(Fig. 3).
Cytoscape 3.6.1 was used to perform a centrality
analysis. The top 15 genes with the highest degree of connectivity
were defined as hub genes. Under this criterion, 15 hub genes were
obtained for the upregulated DEGs, including cyclin-dependent
kinase 1 (CDK1), cyclin B2 (CCNB2), kinesin family
member 2C (KIF2C), topoisomerase (DNA) IIα (TOP2A),
centrosomal protein 55 (CEP55), ribonucleotide reductase
regulatory subunit M2 (RRM2), ubiquitin conjugating enzyme
E2 C (UBE2C), baculoviral IAP repeat containing 5
(BIRC5), centromere protein F (CENPF), NIMA-related
kinase 2 (NEK2), forkhead box M1 (FOXM1), marker of
proliferation Ki-67 (MKI67), protein regulator of
cytokinesis 1 (PRC1), integrin subunit α2 (ITGA2),
and laminin subunit γ2 (LAMC2). Hub genes for the
downregulated DEGs consisted of kininogen 1 (KNG1),
complement C3 (C3), apolipoprotein A1 (APOA1),
albumin (ALB), fibrinogen α chain (FGA),
apolipoprotein B (APOB), fibrinogen γ chain (FGG),
3-hydroxyacyl CoA dehydrogenase (EHHADH), α2-HS glycoprotein
(AHSG), apolipoprotein A2 (APOA2), complement C4A
(C4A), serpin family A member 1 (SERPINA1),
apolipoprotein E (APOE), serpin family C member 1
(SERPINC1) and acyl-CoA oxidase 1 (ACOX1).
To further verify the differential expression of the
critical hub genes in CCA, we evaluated the expression profiles of
30 hub genes in another 4 datasets. GSE76297 consists of 304
specimens in total, 183 of which were utilized in analysis,
including 91 CCA tumor tissues and 92 CCA non-tumor tissues.
GSE26566 consists of 169 specimens in total, 163 of which were
utilized in analysis, including 104 CCA tissues and 59 surrounding
liver tissues. E-GEOD-32879 consists of 37 specimens in total, 23
of which were utilized in analysis, including 16 iCCA tissues and 7
non-tumor tissues. E-GEOD-45001 consists of 10 pairs of iCCA tumor
tissues and non-tumor tissues, which were all utilized in analysis.
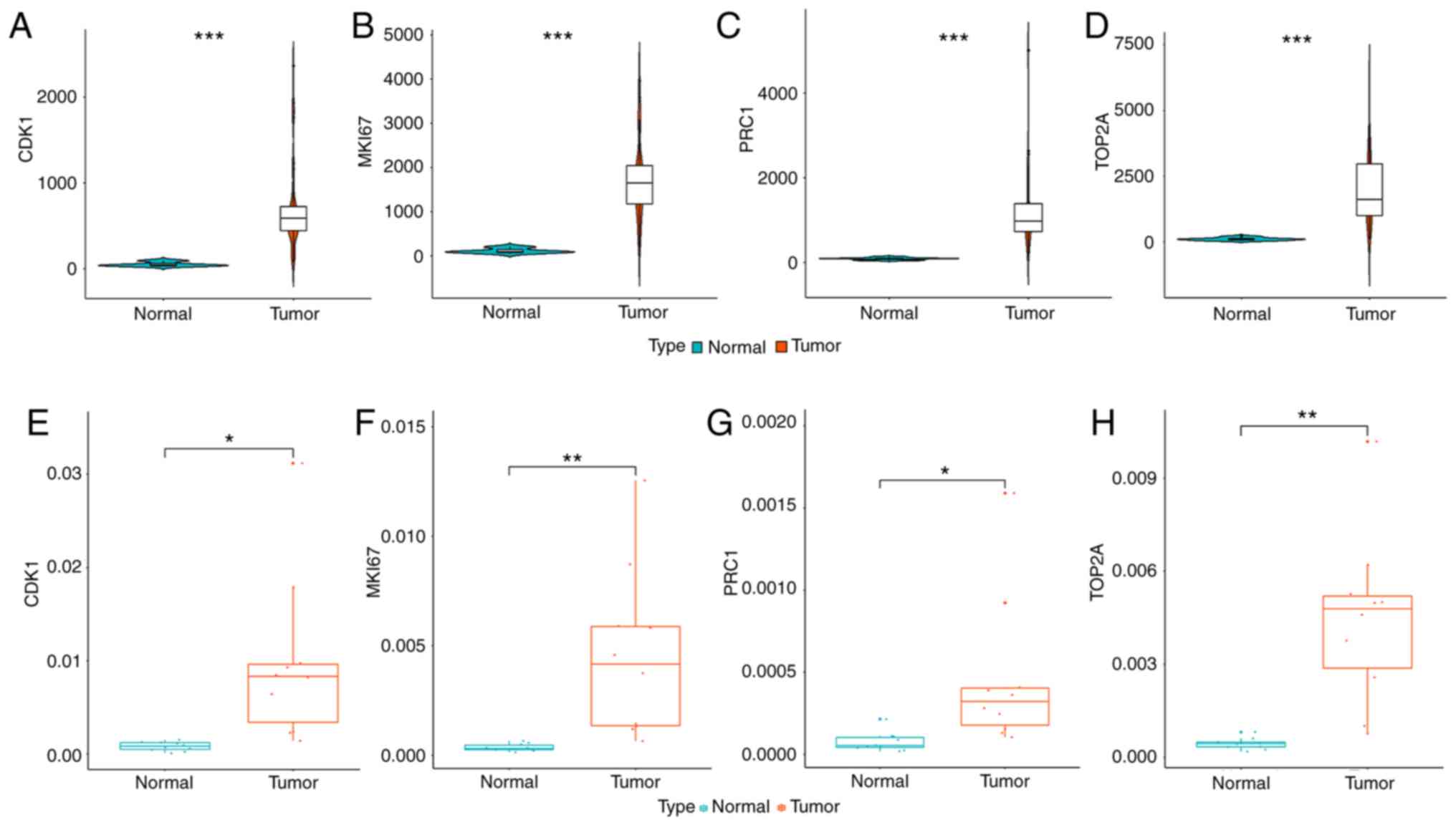
Consistent with our results, 21 out of 30 hub genes in TCGA were
found to share similar differential expression among the other 4
datasets, including 8 upregulated hub genes and 13 downregulated
hub genes (Fig. S1). The
differential expression of hub genes is shown in Fig. 4A-D.
ROC curves and tumor staging
correlation analysis
ROC curves for hub genes were generated based on
expression profile of TCGA dataset. AUC was >0.900 for all 21
selected hub genes (Fig. S2). Among
them, the expression of 5 downregulated hub genes, including
ACOX1, APOA2, APOB, FGA and FGG, was inversely
associated with tumor stage (P<0.05, Fig. S3). For other identified hub genes, no
association between gene expression and tumor stage was found.
Survival analysis
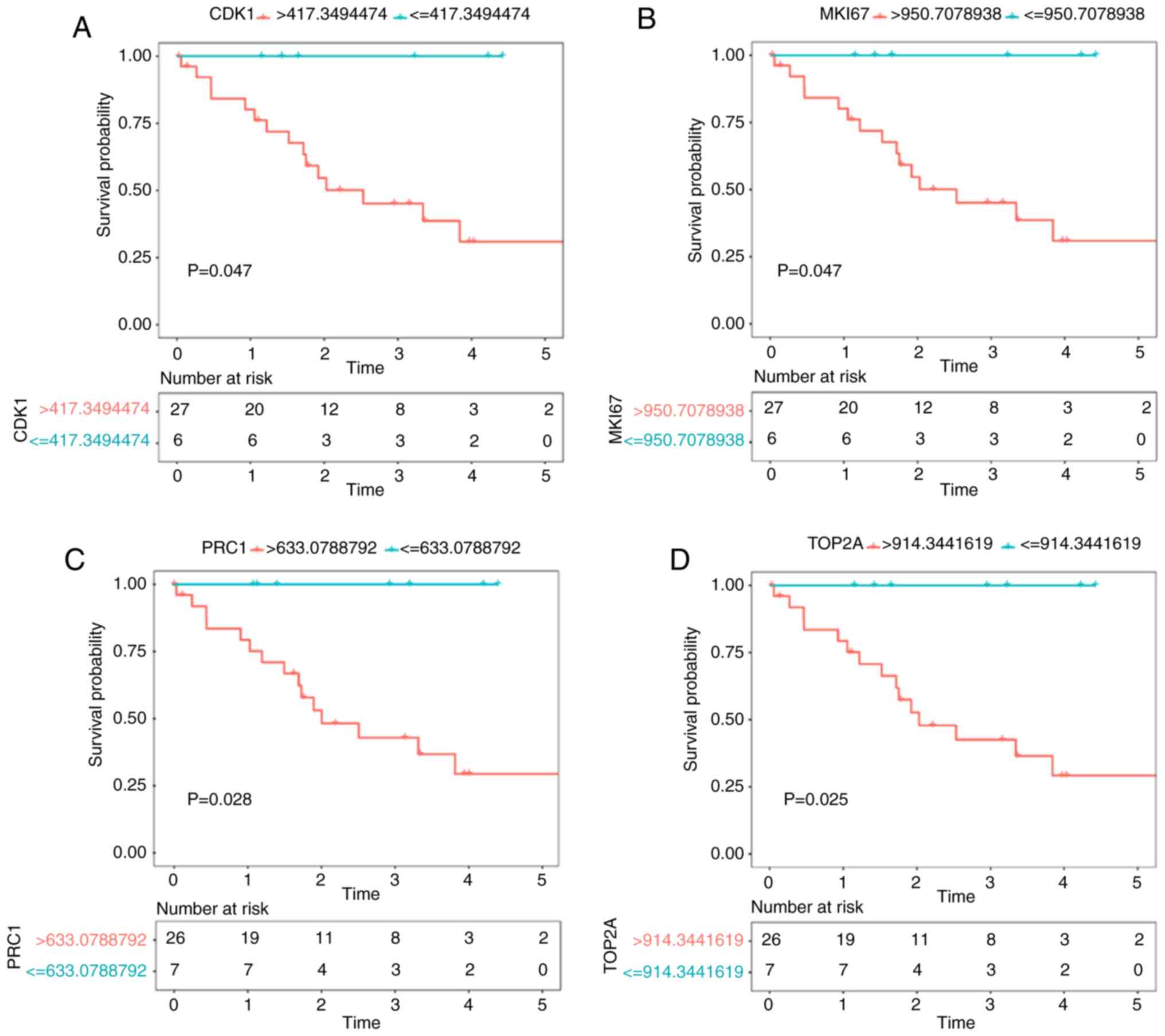
For upregulated hub genes, the expression of
CDK1, MKI67, TOP2A and PRC1 was negatively related to
the overall survival time of CCA patients in both TCGA and GSE89749
datasets (P<0.05). No significant result was found for the
downregulated hub genes. The survival curves are shown in Fig. 5.
Identification of CDK1, MKI67, TOP2A
and PRC1 by RT-qPCR
Since the survival analysis indicates that the
overexpression of CDK1, MKI67, TOP2A and PRC1
predicts poor survival of patients with cholangiocarcinoma, we
performed RT-qPCR to validate the expression change of these genes
in frozen tissue. As expected, all of the 4 genes were upregulated
in the iCCA tissue (Fig. 4E-H).
Discussion
Cholangiocarcinoma (CCA) is recognized as the second
most commonly diagnosed primary liver tumor. Due to its strong
genetic heterogeneity, the current understanding of the
pathogenesis of CCA is not comprehensive. Concerning genetic
changes involved in CCA initiation and progression, agreement in
this field remains fragmented. The key drivers involved in CCA
carcinogenesis still need to be defined (3,4,11). In the present study, we focused on the
genetic changes in transcription level between intrahepatic CCA
(iCCA) and normal tissue. A total of 1,463 differentially expressed
protein-coding genes (DEGs) were obtained based on data from The
Cancer Genome Atlas (TCGA). Gene Ontology (GO) enrichment analyses
showed that ‘changes in cadherin binding involved in ‘cell-cell
adhesion’, ‘extracellular matrix organization’ and the ‘cell-cell
adherens junction’ represented significant GO terms for the
upregulated DEGs and that ‘oxidation-reduction processes’,
‘extracellular exosomes’, and ‘blood microparticles’ represented
significant GO terms for the downregulated DEGs. In addition,
‘ECM-receptor interactions’, ‘focal adhesions’ and ‘small cell lung
cancer’ were significant pathways related to the upregulated DEGs.
‘Metabolic pathways’, ‘complement and coagulation cascades’ and
‘biosynthesis of antibiotics’ were significant pathways for the
downregulated DEGs.
Hub genes were identified based on the degree of
connectivity. Fifteen upregulated hub genes and 15 downregulated
hub genes were selected based on protein-protein interaction (PPI)
network. Moreover, the expression profiles of the 30 hub genes were
verified using datasets from GEO and Arrayexpress. A total of 21
hub genes showed stable differential expression among 5 datasets
including TCGA. ROC curves revealed that all 21 hub genes presented
a credible classification effect between tumor and normal tissue
with AUC >0.900. In addition, the expression of ACOX1, APOA2,
APOB, FGA and FGG was inversely associated with tumor
stage, which indicates that these genes may be involved in the
progression of CCA.
To further explore the relationships between hub
genes and the outcomes of CCA patients, a survival analysis was
conducted based on the clinical data and expression profiles of the
identified hub genes in both TCGA and GSE89749. Four upregulated
hub genes, including CDK1, MKI67, TOP2A and PRC1,
were identified as being significantly related to overall survival
among CCA patients. Moreover, the differential expression of the
four genes was validated by RT-qPCR. Therefore, we considered CDK1,
MKI67, TOP2A and PRC1 as potential predictors for the poor
prognosis of patients with CCA.
Cyclin-dependent kinases (CDKs) are a family of
protein kinases driving the major events of cell cycle control
(29). Aberrant expression of CDK1 is
involved in cell cycle arrest in many tumor types such as melanoma,
colon cancer and pancreatic cancer (30). Studies that have focused on the roles
of CDK1 in cholangiocarcinoma are limited. Okumura et al
revealed that CDK1 is upregulated by AIB1, i.e., transcriptional
coactivator amplified in breast cancer 1, through the Akt pathway.
AIB1 was found to be overexpressed in human CCA specimens and
promote cell cycle progression at the G2/M phase by inducing CDK1
(31). In addition, CDK1 may be
involved in drug-resistant mechanisms of CCA since western blot
analyses indicated that G2/M phase-regulated proteins, including
CDK1, were downregulated in gemcitabine-resistant CCA cell lines
(32).
MK167 encodes Ki-67. It is a cell cycle-regulated
phosphatase 1-binding protein universally used as a proliferation
marker. Ki-67 is a major organizer required for assembly of the
perichromosomal compartment in cells (33). The Ki-67 index is shown to be the most
reliable prognostic evaluation factor of gastroenteropancreatic
neuroendocrine neoplasms (GEP-NENs) (34). The Ki-67 index can be variable through
the disease course (35–37). In combination with tumor type, site
and stage, the Ki-67 index is used to stratify patients in
different prognostic categories (34). In the present study, we found a
prognostic role of MKI67 in CCA. This finding may be clinically
valuable, although the underlying mechanisms for Ki-67 variation
still requires further investigation.
DNA topoisomerase IIα (TOP2A) is an isoform of DNA
topoisomerase II (Topo II). Topo II is a crucial enzyme for cell
division that generates torsional stress on double-stranded DNA by
inducing transient breaks that are subsequently resealed (38). TOP2A is located adjacent to the HER2
oncogene and is frequently coamplified with HER2 in multiple types
of cancers, such as breast cancer, bladder cancer and gastric
adenocarcinoma (39–42) However, Panvichian et al
reported that TOP2A overexpression in hepatocellular carcinoma
(HCC) is independent of HER2 gene amplification or expression
(43). Our results showed that TOP2A
is significantly upregulated in CCA tissues and represents a
possible predictive biomarker for poor prognosis. However, no
significant change was detected in the transcription of HER2.
Therefore, TOP2A may play a role in CCA tumorigenesis independent
of HER2. Nateewattana et al reported that andrographolide, a
Topo II inhibitor, exhibited a potent cytotoxic effect on CCA cells
by suppressing TOP2A expression in vitro (44). Thus, the therapeutic efficacy of Topo
II inhibitors, such as andrographolide and anthracycline, in CCA
patients should be further explored.
Polycomb repressive complex 1 (PRC1) is required for
adult stem cell functions and acts as both a tumor suppressor and
oncogene (45). Tang et al
demonstrated the significant biological implications of PRC1 in
tumor pathogenesis and prognosis in non-small cell lung cancer
patients by analyzing genome-wide RNAi data and mRNA expression
data (46). Bmi1 and EZH2 are
representative members of PRC1. Sasaki et al found that Bmi1
was overexpressed in CCA cell lines and stimulated cell
proliferation (47). Overexpression
of EZH2 may induce hypermethylation of the p16INK4a
promoter, followed by decreased expression of p16INK4a
in multistep cholangiocarcinogenesis (48). However, the precise molecular
mechanisms underlying the role of PRC1 in CCA remain unclear.
Importantly, we noted that both CDK1 and PRC1 were
involved in the same GO term, midbody. Midbody is a transient
structure during cytokinesis and is involved in recruitment and
organization of abscission machinery, which physically regulates
the localization of two daughter cells (49). Midbody dysregulation causes mitotic
problems in daughter cell separation, which increases cancer
susceptibility and tumorigenesis (50). CITRON, a known serine kinase present
at midbody during cytokinesis, could contribute to tumor occurrence
in HCC (51). CDK1 phosphorylates
septin 9 (SEPT9), thus playing an important role in mediating the
final separation of daughter cells (52). Moreover, PCR1 could accumulate in the
midbody during cytokinesis and organize the midbody through
microtubule regulation (49). Based
on this study, we may speculate that CDK1 and PRC1 contribute to
the progression of CCA through midbody-related function.
Although we cannot exclude the possibility that the
identified hub genes may be implicated in noncarcinogenic aspects
of CCA, we attempted to ensure the credibility of the results by
including as many datasets as possible. Except for TCGA, a total of
4 datasets were used to validate the differential expression of hub
genes. In addition, the survival analysis of certain hub genes was
based on 2 datasets. Moreover, we performed RT-qPCR to verify the
selected hub genes based on 10 pairs of tissue samples.
In conclusion, we identified a number of hub genes
and comprehensively revealed the biological functions and signaling
pathways associated with CCA carcinogenesis through systematic
bioinformatic analyses. Moreover, we identified CDK1, MKI67,
TOP2A and PRC1 as possible prognostic biomarkers and
further discussed the roles that the four genes may play in cancer
development. Most of the genes have not been thoroughly studied in
CCA. In future research, the clinical application of the identified
hub genes as biomarkers for supervising the prognosis of CCA
patients should be further investigated. Moreover, research
concerning specific mechanisms of these genes in CCA occurrence and
progression is warranted.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This research was funded by the Beijing Municipal
Science & Technology Commission Research Fund (no.
Z171100000417004) and Beijing Natural Science Foundation (no.
L172055).
Availability of data and materials
Data for CCA mRNA expression were downloaded from
TCGA database (https://portal.gdc.cancer.gov/, RNA-seq, Illumina),
Gene Expression Omnibus (GEO) database of the National Center for
Biotechnology Information (GSE26566, GSE76297), and ArrayExpress
Archive of Functional Genomics Data (E-GEOD-32879 and
E-GEOD-45001). Clinical data of 118 patients with
cholangiocarcinoma were downloaded from PubMed Central (PMID:
28667006). Matching sample information was obtained from GEO
dataset: GSE89749. CCA tissue samples were collected from 10
patients with iCCA undergoing surgery at Peking Union Medical
College Hospital. All patients were enrolled from November 2018 to
April 2019.
Authors' contributions
HL, JuL and YZ designed this study. HL, JuL, FX, KK,
YS, WX, XW, JiL, HX, SD, YX, HZ, YZ, and JG contributed to data
curation and analysis. HL and WX conducted the laboratory
experiment. HL, YS and XW wrote the manuscript. JuL and YZ revised
the manuscript. All the authors read and approved the final version
of the manuscript for publication and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
The study was approved by the Clinical Research
Ethics Committee of Peking Union Medical College Hospital. Each
patient provided written signed informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CCA
|
cholangiocarcinoma
|
|
TCGA
|
The Cancer Genome Atlas
|
|
DEGs
|
differentially expressed
protein-coding genes
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
PPI
|
protein-protein interaction
|
|
GEO
|
Gene Expression Omnibus
|
References
|
1
|
Razumilava N and Gores GJ:
Cholangiocarcinoma. Lancet. 383:2168–2179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang H, Shen F, Han J, Shen YN, Xie GQ,
Wu MC and Yang T: Epidemiology and surgical management of
intrahepatic cholangiocarcinoma. Hepat Oncol. 3:83–91. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bergquist A and von Seth E: Epidemiology
of cholangiocarcinoma. Best Pract Res Clin Gastroenterol.
29:221–232. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu J and Yin B: Advances in biomarkers of
biliary tract cancers. Biomed Pharmacother. 81:128–135. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Esnaola NF, Meyer JE, Karachristos A,
Maranki JL, Camp ER and Denlinger CS: Evaluation and management of
intrahepatic and extrahepatic cholangiocarcinoma. Cancer.
122:1349–1369. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Squadroni M, Tondulli L, Gatta G, Mosconi
S, Beretta G and Labianca R: Cholangiocarcinoma. Crit Rev Oncol
Hematol. 116:11–31. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu AX: Future directions in the treatment
of cholangiocarcinoma. Best Pract Res Clin Gastroenterol.
29:355–361. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rizvi S and Gores GJ: Emerging molecular
therapeutic targets for cholangiocarcinoma. J Hepatol. 67:632–644.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mertens JC, Rizvi S and Gores GJ:
Targeting cholangiocarcinoma. Biochim Biophys Acta Mol Basis Dis.
1864:1454–1460. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ghouri YA, Mian I and Blechacz B: Cancer
review: Cholangiocarcinoma. J Carcinog. 14:12015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jusakul A, Cutcutache I, Yong CH, Lim JQ,
Huang MN, Padmanabhan N, Nellore V, Kongpetch S, Ng AWT, Ng LM,
Choo SP, et al: Whole-genome and epigenomic landscapes of
etiologically distinct subtypes of cholangiocarcinoma. Cancer
Discov. 7:1116–1135. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tomczak K, Czerwińska P and Wiznerowicz M:
The cancer genome atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19:A68–A77. 2015.PubMed/NCBI
|
|
13
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43((Database Issue)): D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng Y, Long J, Wu L, Zhang H, Li L,
Zheng Y, Wang A, Lin J, Yang X, Sang X, et al: Identification of
hub genes involved in the development of hepatocellular carcinoma
by transcriptome sequencing. Oncotarget. 8:60358–60367.
2017.PubMed/NCBI
|
|
15
|
Wang X, Hu KB, Zhang YQ, Yang CJ and Yao
HH: Comprehensive analysis of aberrantly expressed profiles of
lncRNAs, miRNAs and mRNAs with associated ceRNA network in
cholangiocarcinoma. Cancer Biomarkers. 23:549–559. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Anders S and Huber W: Differential
expression analysis for sequence count data. Genome Biol.
11:R1062010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Team RDC: R: A language and environment
for statistical computing. Journal. 2010.
|
|
18
|
Walter W, Sánchez-Cabo F and Ricote M:
GOplot: An R package for visually combining expression data with
functional analysis. Bioinformatics. 31:2912–2914. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Choi JK, Yu U, Yoo OJ and Kim S:
Differential coexpression analysis using microarray data and its
application to human cancer. Bioinformatics. 21:4348–4355. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chaisaingmongkol J, Budhu A, Dang H,
Rabibhadana S, Pupacdi B, Kwon SM, Forgues M, Pomyen Y,
Bhudhisawasdi V, Lertprasertsuke N, et al: Common molecular
subtypes among asian hepatocellular carcinoma and
cholangiocarcinoma. Cancer Cell. 32:57–70.e53. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Andersen JB, Spee B, Blechacz BR, Avital
I, Komuta M, Barbour A, Conner EA, Gillen MC, Roskams T, Roberts
LR, et al: Genomic and genetic characterization of
cholangiocarcinoma identifies therapeutic targets for tyrosine
kinase inhibitors. Gastroenterology. 142:1021–1031.e15. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Oishi N, Kumar MR, Roessler S, Ji J,
Forgues M, Budhu A, Zhao X, Andersen JB, Ye QH, Jia HL, et al:
Transcriptomic profiling reveals hepatic stem-like gene signatures
and interplay of miR-200c and epithelial-mesenchymal transition in
intrahepatic cholangiocarcinoma. Hepatology. 56:1792–1803. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sulpice L, Rayar M, Desille M, Turlin B,
Fautrel A, Boucher E, Llamas-Gutierrez F, Meunier B, Boudjema K,
Clément B and Coulouarn C: Molecular profiling of stroma identifies
osteopontin as an independent predictor of poor prognosis in
intrahepatic cholangiocarcinoma. Hepatology. 58:1992–2000. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sulpice L, Desille M, Turlin B, Fautrel A,
Boudjema K, Clément B and Coulouarn C: Gene expression profiling of
the tumor microenvironment in human intrahepatic
cholangiocarcinoma. Genom Data. 7:229–232. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Robin X, Turck N, Hainard A, Tiberti N,
Lisacek F, Sanchez JC and Müller M: pROC: An open-source package
for R and S+ to analyze and compare ROC curves. BMC Bioinformatics.
12:772011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Therneau TM: A package for survival
analysis in S. version 2.38. 2015, https://CRAN.R-project.org/package=survival
|
|
28
|
Terry M and Therneau PMG: Modeling
Survival data: Extending the Cox ModelDietz K, Gail M, Krickeberg
K, Samet J and Tsiatis A: Springer; New York, NY: 2000
|
|
29
|
Holt LJ, Tuch BB, Villen J, Johnson AD,
Gygi SP and Morgan DO: Global analysis of Cdk1 substrate
phosphorylation sites provides insights into evolution. Science.
325:1682–1686. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ravindran Menon D, Luo Y, Arcaroli JJ, Liu
S, KrishnanKutty LN, Osborne DG, Li Y, Samson JM, Bagby S, Tan AC,
et al: CDK1 Interacts with Sox2 and promotes tumor initiation in
human melanoma. Cancer Res. 78:6561–6574. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Okumura E, Fukuhara T, Yoshida H, Hanada
Si S, Kozutsumi R, Mori M, Tachibana K and Kishimoto T: Akt
inhibits Myt1 in the signalling pathway that leads to meiotic
G2/M-phase transition. Nat Cell Biol. 4:111–116. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wattanawongdon W, Hahnvajanawong C, Namwat
N, Kanchanawat S, Boonmars T, Jearanaikoon P, Leelayuwat C,
Techasen A and Seubwai W: Establishment and characterization of
gemcitabine-resistant human cholangiocarcinoma cell lines with
multidrug resistance and enhanced invasiveness. Int J Oncol.
47:398–410. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Booth DG, Takagi M, Sanchez-Pulido L,
Petfalski E, Vargiu G, Samejima K, Imamoto N, Ponting CP, Tollervey
D, Earnshaw WC and Vagnarelli P: Ki-67 is a PP1-interacting protein
that organises the mitotic chromosome periphery. Elife.
3:e016412014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Klöppel G and La Rosa S: Ki67 labeling
index: Assessment and prognostic role in gastroenteropancreatic
neuroendocrine neoplasms. Virchows Arch. 472:341–349. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Miller HC, Drymousis P, Flora R, Goldin R,
Spalding D and Frilling A: Role of Ki-67 proliferation index in the
assessment of patients with neuroendocrine neoplasias regarding the
stage of disease. World J Surg. 38:1353–1361. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Singh S, Hallet J, Rowsell C and Law CHL:
Variability of Ki67 labeling index in multiple neuroendocrine
tumors specimens over the course of the disease. Eur J Surg Oncol.
40:1517–1522. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Grillo F, Albertelli M, Brisigotti MP,
Borra T, Boschetti M, Fiocca R, Ferone D and Mastracci L: Grade
increases in gastroenteropancreatic neuroendocrine tumor metastases
compared to the primary tumor. Neuroendocrinology. 103:452–459.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Romero A, Martín M, Cheang MC, López
García-Asenjo JA, Oliva B, He X, de la Hoya M, García Sáenz JÁ,
Arroyo Fernández M, Díaz Rubio E, et al: Assessment of
Topoisomerase II α status in breast cancer by quantitative PCR,
gene expression microarrays, immunohistochemistry, and fluorescence
in situ hybridization. Am J Pathol. 178:1453–1460. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Järvinen TA and Liu LE: Simultaneous
amplification of HER-2 (ERBB2) and topoisomerase IIalpha (TOP2A)
genes-molecular basis for combination chemotherapy in cancer. Curr
Cancer Drug Targets. 6:579–602. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liang Z, Zeng X, Gao J, Wu S, Wang P, Shi
X, Zhang J and Liu T: Analysis of EGFR, HER2, and TOP2A gene status
and chromosomal polysomy in gastric adenocarcinoma from Chinese
patients. BMC Cancer. 8:3632008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Press MF, Sauter G, Buyse M, Bernstein L,
Guzman R, Santiago A, Villalobos IE, Eiermann W, Pienkowski T,
Martin M, et al: Alteration of topoisomerase II-alpha gene in human
breast cancer: Association with responsiveness to
anthracycline-based chemotherapy. J Clin Oncol. 29:859–867. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Simon R, Atefy R, Wagner U, Forster T,
Fijan A, Bruderer J, Wilber K, Mihatsch MJ, Gasser T and Sauter G:
HER-2 and TOP2A coamplification in urinary bladder cancer. Int J
Cancer. 107:764–772. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Panvichian R, Tantiwetrueangdet A,
Angkathunyakul N and Leelaudomlipi S: TOP2A amplification and
overexpression in hepatocellular carcinoma tissues. Biomed Res Int.
2015:3816022015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nateewattana J, Dutta S, Reabroi S, Saeeng
R, Kasemsook S, Chairoungdua A, Weerachayaphorn J, Wongkham S and
Piyachaturawat P: Induction of apoptosis in cholangiocarcinoma by
an andrographolide analogue is mediated through topoisomerase II
alpha inhibition. Eur J Pharmacol. 723:148–155. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gil J and O'Loghlen A: PRC1 complex
diversity: Where is it taking us? Trends Cell Biol. 24:632–641.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tang H, Xiao G, Behrens C, Schiller J,
Allen J, Chow CW, Suraokar M, Corvalan A, Mao J, White MA, et al: A
12-gene set predicts survival benefits from adjuvant chemotherapy
in non-small cell lung cancer patients. Clin Cancer Res.
19:1577–1586. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sasaki M, Yamaguchi J, Ikeda H, Itatsu K
and Nakanuma Y: Polycomb group protein Bmi1 is overexpressed and
essential in anchorage-independent colony formation, cell
proliferation and repression of cellular senescence in
cholangiocarcinoma: Tissue and culture studies. Hum Pathol.
40:1723–1730. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sasaki M, Yamaguchi J, Itatsu K, Ikeda H
and Nakanuma Y: Over-expression of polycomb group protein EZH2
relates to decreased expression of p16 INK4a in
cholangiocarcinogenesis in hepatolithiasis. J Pathol. 215:175–183.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hu CK, Coughlin M and Mitchison TJ:
Midbody assembly and its regulation during cytokinesis. Mol Biol
Cell. 23:1024–1034. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sadler JBA, Wenzel DM, Williams LK,
Guindo-Martínez M, Alam SL, Mercader JM, Torrents D, Ullman KS,
Sundquist WI and Martin-Serrano J: A cancer-associated polymorphism
in ESCRT-III disrupts the abscission checkpoint and promotes genome
instability. Proc Natl Acad Sci USA. 115:E8900–E8908. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fu Y, Huang J, Wang KS, Zhang X and Han
ZG: RNA interference targeting CITRON can significantly inhibit the
proliferation of hepatocellular carcinoma cells. Mol Biol Rep.
38:693–702. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Estey MP, Di Ciano-Oliveira C, Froese CD,
Fung KY, Steels JD, Litchfield DW and Trimble WS: Mitotic
regulation of SEPT9 protein by cyclin-dependent kinase 1 (Cdk1) and
Pin1 protein is important for the completion of cytokinesis. J Biol
Chem. 288:30075–30086. 2013. View Article : Google Scholar : PubMed/NCBI
|