Chimeric antigen receptors (CARs) are unique
receptors that are designed to target a specific tumor antigen to
functionally reprogram T lymphocytes. As T lymphocytes are
genetically engineered to express these artificial receptors to
target cancer cells, the type of therapy may be termed
immunotherapy, gene therapy or cancer therapy (1). The human defense system can efficiently
identify self and non-self molecules, including bacteria, viruses
and abnormal cancer cells. The identification of tumor cells is
based on their acquired antigenicity and immunogenicity via the
expression of foreign antigens (2).
However, cancer cells have the potential to subvert the immune
system to their advantage, resulting in inadequate antitumor
immunity, and tumor survival and progression (3). Immunotherapy is also termed biotherapy
as the immune system in the body is naturally capable of detecting
pathogens and cancerous cells. In recent years, immunotherapy has
emerged as an important branch of treatment for similar types of
disease; however, its protective mechanism may differ (4). Certain immunotherapies boost the immune
system, whereas others directly target the cancer cells. Each
treatment type has its advantages and disadvantages depending on
the disease type (5).
Tisagenlecleucel (Kymriah) is a medication used to treat patients
with acute lymphoblastic leukemia (ALL) up to the age of 25 years.
Similarly, axicabtagene ciloleucel (Yescarta) is approved for
patients with large B-cell lymphoma, such as non-Hodgkin lymphoma
(NHL), and those with cancer in a refractory and recurrent state or
whose cancer is non-responsive to other treatments (6). With increasing awareness of the immune
system, a number of innovative immunotherapies are being developed
using methods which include inducing the immune system to function
in an impertinent manner to target malignant cells. Another
approach involves the administration of immune components, such as
synthetic, modified immune proteins that are genetically engineered
to target tumor antigens (7).
CAR T cell treatment has achieved success in
treating hematopoietic malignancies; however, its effectiveness
against solid tumors remains to be determined. In the following
sections, the rapid progression in implementing the adoptive
transfer of T cells and their mechanism of tumor cell eradication
are discussed.
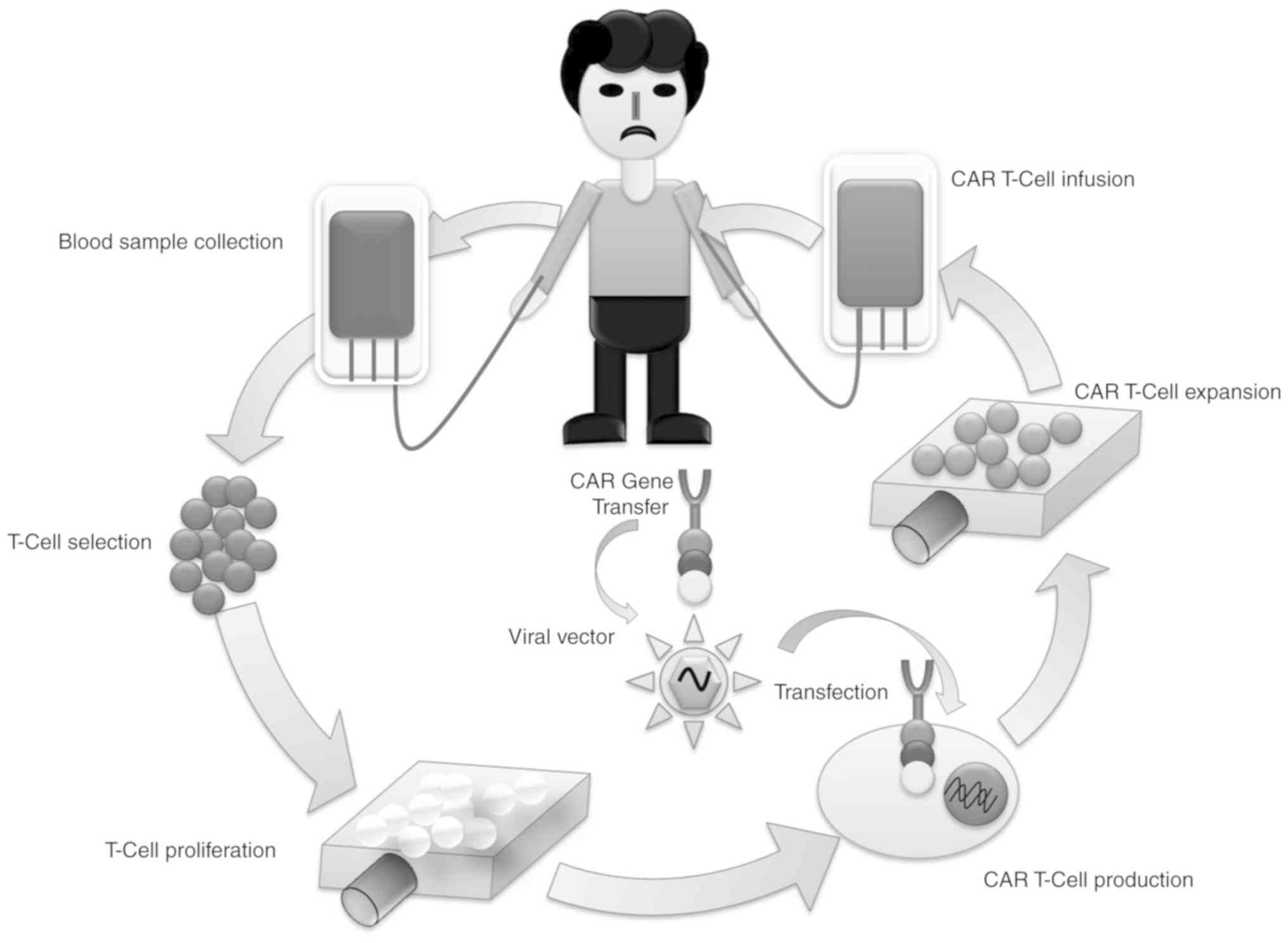
CAR is an emerging immunotherapy for several
malignancies. This therapeutic approach is an experimental form of
gene therapy that redirects T lymphocytes to eradicate cancerous
cells. The initial step in this therapy is leukapheresis or the
isolation of a patient's peripheral blood (8,9). Apheresis
is widely used to isolate blood from patients and separate it into
its components, which are then genetically altered before
re-injecting them into the patient's body. Currently, apheresis is
used by blood banks to collect platelets and other blood components
for the treatment of several diseases, including hematologic and
renal disorders. Therefore, it is regarded as a safe practice for
healthy individuals and patients (10) (Fig.
1).
The uniqueness of chimeric receptors exists in their
ability to fuse or split discrete vital functions, such as
recognition, co-stimulation and activation, in different chains of
a receptor molecule by imitating the complexity of the native T
cell receptor (TCR) structure (11).
T cells do not usually require costimulation for activation and to
initiate proliferation, but in the process of establishing CAR T
cells, the activation and proliferation of T cells require the
presence of costimulatory molecules, which also assist in CAR T
cell cytokine production. The strategy involves constructing an
engineered chimeric receptor for T cells based on the integration
of scFv fragments in the hinge area that separates scFv from the
cell membrane. The exposure of scFv on the cell surface, in
addition to other small functional molecules, enhances induction of
the cytolytic function of the engineered T cell. Together, this
coordination of a ‘living drug’ in the immune system fights against
cancer (12). In addition, CAR T
cells can remain stable for several years in the body as long-term
memory cells. This feature allows them to recognize and kill cancer
cells encountered in the circulation in the case of relapse.
Another advantage of CAR T cells is that they specifically target
only tumor cells and not auto-antigens. Therefore, it is safe and
nonlethal to host cells (13). Once
the synthetic immunoreceptor is expressed on the surface of an
engineered T cell, its scFv specifically binds to target antigens
expressed on a cancer cell. This binding subsequently results in
the transduction of an activating signal into the genetically
edited immune cell. The T cell then elicits its effector anticancer
function (14). CAR T cell therapy is
considered to be the first approach to reconfigure T lymphocytes
with an antibody-specific scFv fragment obtained from monoclonal
antibodies (mAbs) by replacing different parts of the TCRα and β
chains. A recent report stated the potential of CAR γδ T cells in
curing mucosal-derived malignant tumors as a novel strategy for CAR
T cell therapy (15). The hybrid TCR
functionally expresses and recognizes the analogous target antigen
molecules in a non-MHC-restricted manner. Depending on the diverse
biomarker selection and structural complexity, different
generations of CAR model have been developed (16).
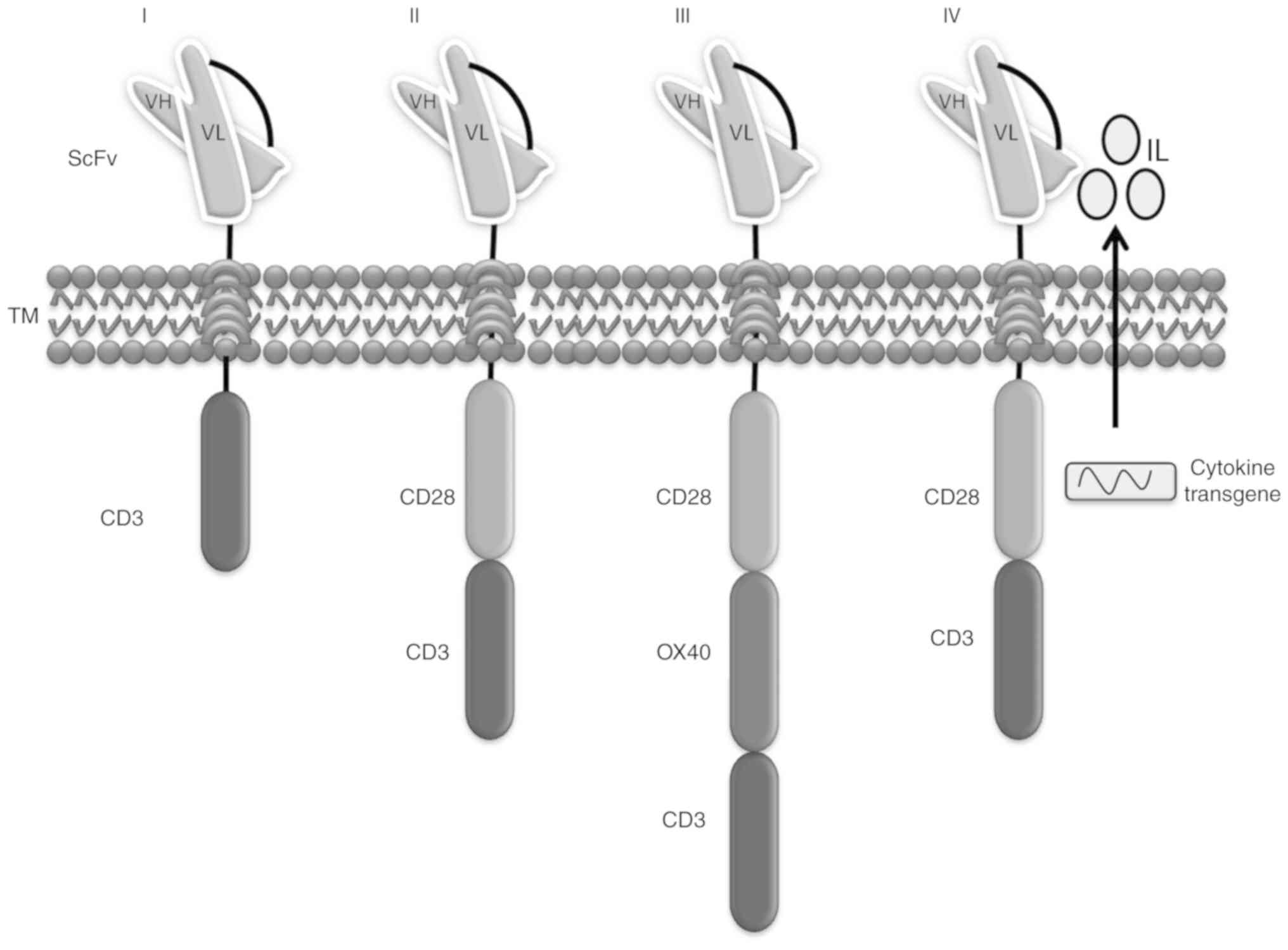
The first-generation CAR T model comprises a CD3ζ
chain as a key transmitter of signals from endogenous TCRs.
Following a successful outcome in pre-clinical trials, this type of
drug entered into phase I clinical trials for leukemia, lymphoma
and various other types of cancer, including ovarian cancer and
neuroblastoma (17). Despite the
inadequate antitumor action owing to the lack of activation,
persistent exposure to the tumor environment has resulted in
continued therapeutic effects in patients with B-cell lymphoma
infused with α-CD20-CD3ζ CAR T cells and a number of patients with
neuroblastoma treated with scFv-CD3ζ CAR T cells (18). The heavy and light chains constitute
structural parts of the B cell receptor or antibodies, called scFv,
which are fused to the CD3 domain or T cell-activating ζ chain of
the TCR to create non-MHC-restricted activating receptor molecules.
These modified molecules are capable of enhancing T cell antigen
detection and cytotoxicity by specifically targeting tumor cells
(19). The original ‘true’ CAR was
designed by integrating an scFv antibody receptor directly with the
CD3ζ domain. This model was subsequently named the ‘T body
approach’ and these synthetic signaling receptors are now known as
CARs or chimeric immune receptors (20).
The success of first-generation CARs in phase 1
clinical trials paved the way for second-generation CAR T cell
therapy. This model of CAR T cells was established to elicit a more
effective anti-leukemic response in phase I clinical trials with
complete remission rates of up to 90% in patients with recurrent
B-cell ALL (B-ALL). Here, the second-generation anti-CD19 T cells
were integrated into a 4-1BB or CD28 co-stimulatory domain attached
to the CD3 domain (21). However,
significant concerns remain regarding its efficacy and safety and
making it more robust. Anticipating these concerns,
second-generation CARs aimed at integrating intracellular signaling
domains from different co-stimulatory molecules, such as CD28,
4-1BB or CD137, inducible T cell costimulator (ICOS) or CD278, OX40
or CD134 fused to the cytoplasmic tail of the CAR, thus amplifying
the signal (22). First-generation
CARs contain a CD3ζ chain as a key transmitter of signals from
endogenous TCRs, whereas second-generation CARs contain a CD3ζ
chain and a single costimulatory molecule, which is why the
receptor is known as a second-generation CAR. For example,
anti-CD19 CARs consisting of 4-1BB or CD28 signaling domains
produced notable complete response (CR) rates in patients with
relapsed and refractory B-cell malignancies (23). CR represents complete response or
disappearance of all signs of cancer in response to treatment [The
United States Food and Drug Administration (FDA) 2007].
Consequently, CD28-based CARs have a rapid proliferative reaction,
thus enhancing T effector cell functions, whereas 4-1BB-based CARs
lead to improved T cell accumulation (24).
To extend the antitumor efficacy, third-generation
CARs comprise two signaling domains and the CD3ζ chain, such as
CD3ζ-CD28-OX40 and CD3ζ-CD28-4-1BB, to achieve improved activation
signal, prolonged proliferation, elevated cytokine production and
effective function (25). For
example, a third-generation CAR consisting of
α-CD19-CD3ζ-CD28-4-1BB reported complete remission rates by
infiltrating and lysing cancer tissue in patients with chronic
lymphocyte leukemia (26). In
addition, certain CAR T cells function as memory cells, thus
preventing tumor relapse. Despite the significant curative effect,
the irrepressible activity of CARs and their increased antitumor
efficacy is associated with life-threatening and unfavorable
outcomes, with increased secretion of pro-inflammatory cytokines,
multi-organ dysfunction, pulmonary failure and death (27).
All earlier CARs were based on a precise stratagem
and helped in mediating the T cell anticancer response. However,
these suffered from limitations, including a lack of antitumor
activity against solid tumors owing to large phenotypic
heterogeneity and deterioration attributed to antigen-negative
cancer cells. These shortcomings led to the development of a novel
CAR stratagem (28). The
fourth-generation CAR was introduced to establish the tumor
background via the inducible expression of transgenic immune
modifiers, such as interleukin (IL)-12, which activate innate
immune cells and enhance T cell activation to reduce
antigen-negative cancer cells in the marked lesion (29,30)
(Fig. 2).
Gene therapy involves the delivery of DNA into cells
and this can be accomplished using a number of methods summarized
below. The most traditional method utilizes recombinant viruses
(also known as viral vectors), biological nanoparticles and
non-viral methods based on the direct delivery of naked DNA.
Viral vectors, such as γ-retrovirus, lentivirus and
adenovirus vectors are generally used in gene therapy. Retrovirus
transduction is one of the commonly used delivery methods in gene
therapy. The method involves a reverse transcriptase that promotes
the stable integration of artificial genes into the host genome
(31). To create a vector,
γ-retroviral coding sequences are substituted by a gene of
interest. The retroviral vectors possess an innate capability to
disturb the genomic section and results in neoplastic
transformation. Thus, γ-retroviral vectors have been used in gene
therapy applications (32).
Lentiviral vectors are retroviruses derived from human
immunodeficiency virus-1, which serve as a major tool for the
delivery of transgenes into mammalian cells. The advantage of using
lentivirus vectors is their efficient transduction and their stable
integration and expression into non-dividing and dividing cells
both in vitro and in vivo (33).
The mechanism of transposon transfection is
different from that of viral transfection. Delivery via transposons
is a non-viral process that uses transposon DNA and a transposase
enzyme for stable gene transfer. Two important vectors, namely
piggyBac (PB) and sleeping beauty (SB), are frequently used
(34). Transposons shift from one
gene position to another position via a cut-and-paste mechanism.
The mechanism of the transposon system using SB and PB involves
four steps: i) The transposase enzyme helps in recognition and
binding to the transposon; ii) a synaptic complex is produced by
the coupling and binding of the repeat elements at both ends of the
transposon; iii) the transposon excises the genetic element to be
transposed, and iv) the excised element is reintegrated into the
target location (35). The
transposase for both vectors (SB and PB) consists of the
DNA-binding domain, transposable catalytic domain and nuclear
localization signal. The SB vector is a safer substitute for viral
vectors owing to its innately low enhancer activity, non-pathogenic
source and minimum epigenetic alterations at the incorporation site
(36). By contrast, the PB vector has
been shown to provide a large load capacity and additional
competent transposition action in in vivo studies. PB
vector-mediated CAR T therapy is considered safer than that based
on the SB vector. Therefore, use of the PB vector results in
greater CAR production, as evident from the production of CAR
targeting CD19 using a PB vector (37).
Electroporation has evolved as a useful technique
for modifying genes of diverse cell types. The target cells are
exposed to electric fields to temporarily disrupt their cell
membranes. This allows the charged molecules to enter the cells.
Square-wave and pulse-based systems are new electroporation devices
(38). The Lonza Nucleofector II
Electroporator performs effective genetic alteration of T cells
using certain electric parameters and electroporation buffers
(39). The electroporation of human T
cells has been reported to be associated with ~40–60% of gene
expression and 80% of cell viability (40). One of the limitations of
electroporation is the low transfection efficiency and redundant
cell damage (41).
The most critical step in CAR T cell therapy is T
cell activation, for which cells need to be incubated with the
viral vector including a CAR gene. However, stable insertional
mutagenesis can be dangerous as its possible effects on humans
remain to be fully elucidated (42).
Retroviral or lentiviral vectors offer permanent gene integration
into host cells, with the potential integration in close proximity
with a proto-oncogene which can result in an oncogene. The various
disadvantages associated with these transfer methods suggest that a
safer alternative is needed (43).
Although CAR therapy has emerged as a promising
anticancer approach, it is not free from challenges that require
optimization. For example, the enhanced persistence and improved
cytotoxic profile of CAR T cell therapy are active areas of
research requiring long-term follow-up in clinical trials (48). In addition, a number of serious side
effects have been known to be frequently associated with CAR T cell
therapy, including neurological toxicity, cytokine release syndrome
(CRS), B cell aplasia, tumor lysis syndrome and anaphylaxis
(49). The proliferation of CAR T
cells produces cytokines in the body which kill cancer cells. The
symptoms of CRS-associated toxicity range from mild symptoms of
fatigue, nausea, headache, fever and chills to serious symptoms
including a lowering of blood pressure, tachycardia and capillary
leakage. Another side effect is the presence of CAR T cells
targeting antigens on the surface of B cells or T cells that not
only target cancer cells but also normal cells (50), resulting in B cell aplasia. Therefore,
a thorough investigation is essential to measure the properties of
B cell aplasia. Similarly, tumor lysis syndrome can result in
toxicity by the collapse of dead cells generally in the beginning
of cancer treatment. It could also cause organ damage and be
life-threatening to the patient (51).
Although cancer cells have multiple lineages and
heterogeneity, they possess common target antigens, such as CD19,
CD20, CD22 and numerous others that allow CAR T cells to recognize
tumor cells irrespective of cell lineage. Therefore, recent
advances in this technique include more precise target antigens
expressed by tumor cells (13,52). This
review places additional emphasis on new studies of CAR T cell
therapy that distinguish diverse CAR T cells which can enhance
tumor cell death. Certain models are already being implemented and
others are in the clinical investigation status, including NK-CAR
and clustered regularly interspaced short palindromic repeats
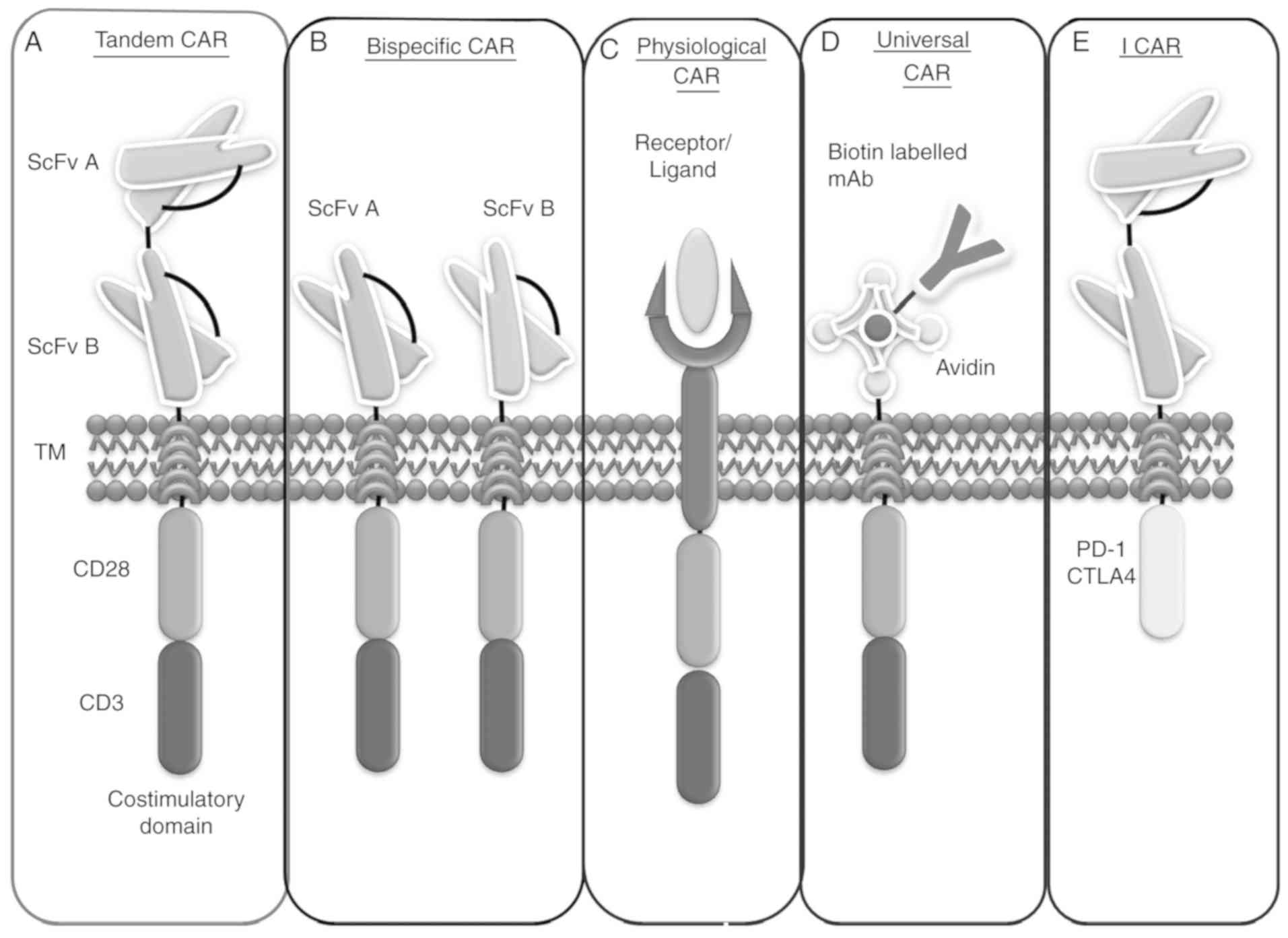
(CRISPR)-CAR therapy (Fig. 3A-E).
A bispecific receptor is one that contains two
distinct antigen recognition domains attached and placed with two
distinct intracellular signaling domains that are expressed as two
different CARs on a single cell surface. At present, bispecific CAR
CD19/CD20 has been introduced as a novel synthetic molecule that
can recognize and bind to more than one targeted tumor antigen on
the cancer cell surface. Therefore, it can create a synergistic
cascade of effector molecules when it encounters two tumor antigens
(53). Additionally, the bispecific
CAR conserves the cytolytic capacity of T cells, i.e., if one of
the objective molecules is not accessible to CAR T cells due to a
cellular hindrance such as mutation of a target antigen or loss of
the target antigen commonly found in malignant cells, a bispecific
CAR can counterbalance the tumor evasion (54). Investigated therapeutics include CD3
of T cells and tumor antigens, such as CD19, on malignant cells.
The curative ability is evident from blinatumomab, an approved
bispecific T cell-engager against relapsed/refractory B-ALL
(55). Positive results have been
reported in patients <15 years of age with relapsed/refractory
ALL, with a heightened response in 26/36 (72%) patients in 9
months. Blinatumomab was discovered while studying a patient who
was negative for minimal residual disease (MRD) and who was found
to be MRD-positive following consolidated chemotherapy (56). Numerous other bispecific CARs have
been investigated preclinically by several research organizations,
including CD20/CD19 and CD20/CD3 (57,58).
Targeting T cells with bispecific CARs was shown to eliminate
transplanted pediatric ALL in a study, whereas T cells targeted by
CD20 CAR did not control the disease (59).
Conventional CARs cannot meet higher expectations
under certain circumstances, such as the downregulation or
alteration of targeted antigens that can occur in cancerous cells.
These conditions subsequently lead to antigenic loss or escape
variations (60). To overcome this
shortcoming, scientists are attempting to develop advanced
technology, in which two particular antigen recognition sites are
joined by a linker, placed in tandem on a single intracellular
domain and expressed as a single CAR on a cell surface, termed a
Tan-CAR. This model enables the synchronized targeting of both
antigens on a single cancer cell or tumor microenvironment. In this
manner, it enhances the activation and stimulation of T cells by
increasing their avidity and expanding their therapeutic properties
(61). Tan-CARs have several
significant curative implications as they are as potent as standard
disease models with single antigen-specific CARs. Additionally,
they are more efficient and less noxious in a higher disease load
setting (62). This may be attributed
to optimized cytokine production and limited cell killing, as
evident from preclinical studies on Tan-CAR T cells in a mouse
tumor model, which confirmed its possibility for remedial
implication in human disease consisting of B-cell antigen CD19 and
human epidermal growth factor receptor 2 (HER2) (63). Recently, a trivalent CAR T cell was
designed by a group of scientists by simultaneously co-targeting
multiple antigens, such as HER2, IL-13 receptor α2 and ephrin A2,
to overcome interpatient changeability with a propensity to target
almost 100% of tumor cells (64).
It has been reported that certain novel
immunoinhibitory receptors are involved in T cell activation and
the attenuation or termination of T cell responses, such as
programmed death-1 (PD-1), programmed death-ligand 1 (PD-L1) and
cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4). These
pathways are regarded as cancer immunotherapy breakthroughs
(65). Recently, two immunologists
won the Novel Prize in Physiology/Medicine, Dr James Allison and Dr
Tasuku Honjo, for their profound discovery in the field of cancer
immunotherapy. Dr Allison's work focused on the T cell surface
protein CTLA-4; he found that the inhibition of immune cells and
antibodies against CTL-4 eliminated malignant growth and prevented
new tumor formation (66). This
finding was tested on 14 patients with metastatic melanoma, with
relapse observed in three patients. In 2011, the FDA approved an
anti-CTLA-4 (ipilimumab) antibody as a treatment for high-grade
melanoma (67).
Similarly, Dr Honjo's team investigated a novel T
cell protein, PD-1, which was discovered in 1992 (68). They observed that PD-L1 presenting on
healthy cells and malignant cells bind to PD-1. They reported
another similar molecule, PD-L2, that also binds to PD-1. Based on
these findings, they published a report that malignant cells
produced PD-L1 and blocked PD-L1 using a counteracting antigen to
inhibit tumor growth (69). The first
clinical trials to target malignancy were launched in 2006, which
indicated significant viability in a number of patients in 2012.
The FDA approved the main PD-1 checkpoint inhibitors, nivolumab and
pembrolizumab (70,71), for treating melanoma in 2014 (72). However, a single treatment may not be
sufficient, which is the reason that a consolidated treatment
targeting CTLA-4 and PD-1 is currently being investigated. Both Dr
Allison and Dr Honjo encouraged the merging of various strategies
to generate more advanced forms of the drug for the immune system
to inhibit tumor cells more effectively (73).
Certain tumor cells contain a high level of PD-L1,
which assists in evading immune attack. It has already been
investigated that PD-1- or CTLA-4-based I-CARs can efficiently
control the cytotoxicity, secretion of cytokines, and proliferation
and cytotoxicity stimulated by endogenous TCR or an activating
chimeric receptor (74). I-CARs are
designed to control the actions of CAR T cells through inhibitory
receptors. This advanced feature unites the action of two chimeric
receptors; one of these generates a dominant-negative signal that
restricts the responses of activated CAR T cells by the activating
receptor. I-CARs can inhibit the activator CAR response to antigens
expressed only by normal cells, thus differentiating between cancer
and normal cells (75). Therefore,
using genetic engineering to inhibit T cell inhibition physiology
and regulate T cell response can be harnessed in an
antigen-selective manner. This has been experimentally confirmed
preclinically in a mouse model by designing an I-CAR using surface
antigen recognition domains CTLA-4 and PD-1. In mice lacking CTLA-4
receptor, substantial T cell activation and proliferation were
observed, ultimately leading to rigorous systemic autoimmune
disease (76). Similarly, PD-1 is
another the inhibitory receptor, which was found to be particularly
expressed by activated T cells causing glomerulonephritis and
arthritis in C57BL/6 mice and certain non-obese diabetic mice
affected by insulitis (77).
Initially, several CAR constructions contained scFv
of murine origin. This is associated with the risk of an immune
response to the modified cells, and the resulting anaphylaxis of
CAR T cell transfer cannot be avoided. These disadvantages limit
the persistence of the infused cells. Therefore, besides
conventional CAR, a physiological CAR has been developed, also
known as a receptor-ligand CAR, which can recognize and bind to
tumor antigens, such as HER3 and HER4 (78). The physiological CAR consists of an
antigen receptor and a CD3ζ intracellular signaling domain with or
without a transmembrane region, which can also be engineered into
immune cells to target the ligands expressed on tumor cells. This
approach increases the capability of T lymphocytes to distinguish
tumor-related targets and eliminate cancer cells (79). This physiological CAR is an emerging
field of CAR T cell therapy with limited published reports.
Experimental trials may have been initiated but the outcomes have
not yet been published.
Although scFv is specifically directed against
tumor-associated antigens, the recognition specificity potential of
CAR T cells is inadequate. Therefore, uCARs were developed to
overcome this limitation. To construct a universal CAR, biotin or
anti-fluorescein isothiocyanate (FITC) scFv is used as a targeting
region, which is fused to a transmembrane domain with one or two
endodomains (80). The
uCAR-expressing T cells can efficiently recognize and remove cancer
cells through the binding of FITC-labeled or biotinylated
antigen-specific mAbs, which, in turn, activate the T cells and
stimulate their proliferation and production of cytokines (81). The mSA2 CAR T cell is a uCAR, which
can be fused to a biotinylated tumor-specific antibody to
specifically target various types of tumor. As with interferon γ,
it is well equipped for interceding malignant cell lysis and
cytokine production (82). Human
clinical trials involving uCAR T cells are ongoing. Two children
with relapsed and refractory B-ALL achieved molecular and cytogenic
remission following uCAR T therapy. Clinical trials have been
initiated on uCAR T cell therapy specific to CD19-positive cells
(NCT03166878 and NCT03229876). However, detailed information and
conclusions are not yet available (83).
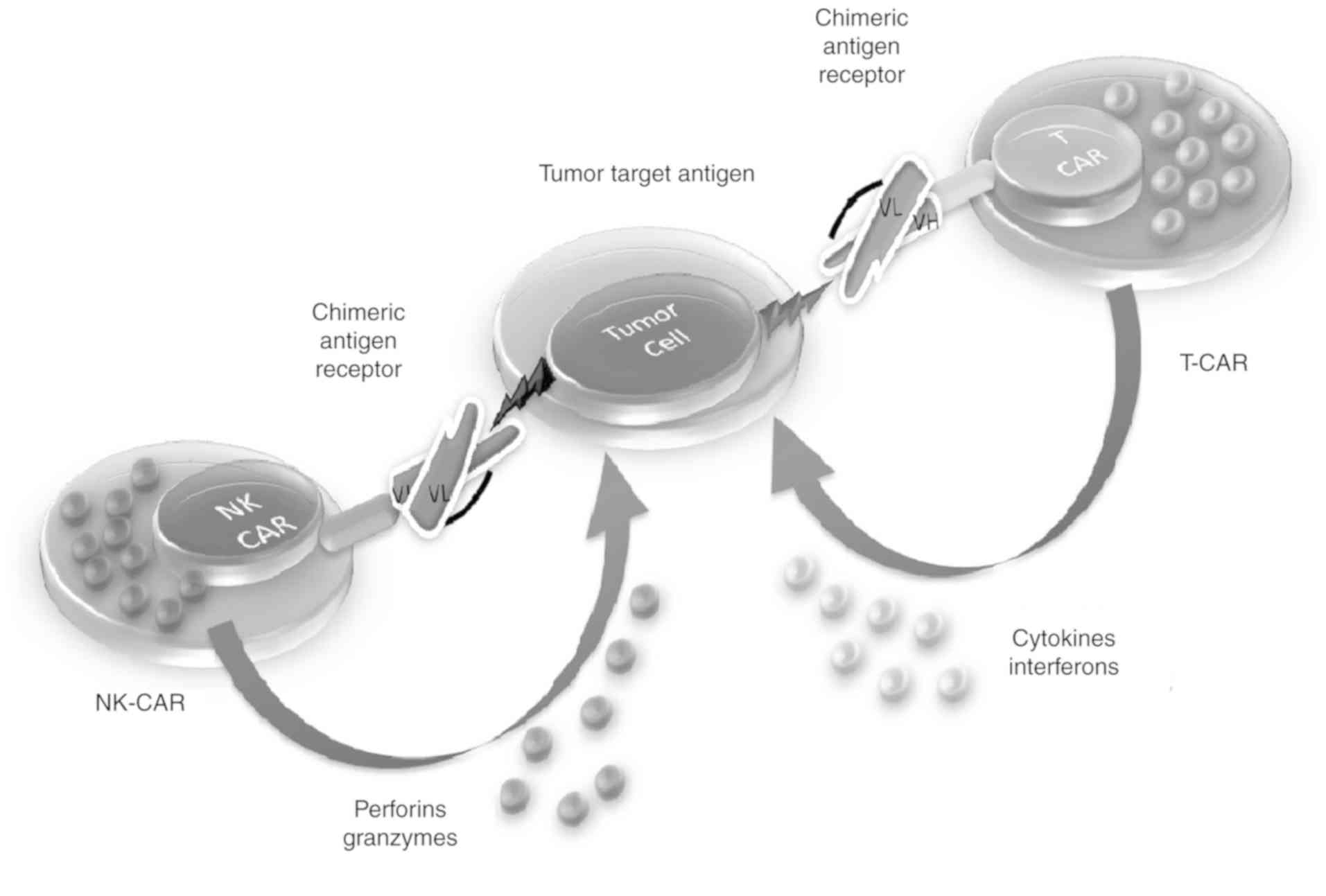
NK cells are a type of cytotoxic T cell that are
essential for natural immunity. The function of NK cells is similar
to that of cytotoxic T cells in the adaptive immune response of
vertebrates. Immune cells generally identify MHC complexes
expressed on infectious cell surfaces to elicit cytokine
production, thus generating an immune response, culminating in the
apoptosis or death of infectious cells (84). NK cells are the only immune cells that
identify infected cells in the absence of MHC and antibodies, thus
eliciting a rapid immune response. These are known as ‘natural
killers’ as they do not require activation to destroy cells that
are devoid of ‘self’ MHC class I molecule markers (85), making them important for destructive
cells with missing MHC I markers.
Cancer cells that do not cause any inflammation are
treated as self by the immune system and do not stimulate a T cell
response. NK cells produce a number of cytokines, including tumor
necrosis factor α, interferon γ and IL-10, which act as immune
suppressors (86). The activation of
NK cells leads to the gradual formation of cytolytic effectors
cells, such as dendritic cells, macrophages and neutrophils, which
consequently facilitate antigen-specific T and B cell responses. NK
cell-mediated tumor cell lysis involves several receptors,
including NKp44, NKp46, NKG2D, NKp30 and DNAM. Malignant cells
usually express NKG2D in addition to ULBP and MICA (87,88). The
clinical effectiveness of CAR T cells has been shown for ALL;
however, this therapeutic approach has not been confirmed for acute
myeloid leukemia (AML), suggesting the need for other therapeutic
options (89). Hypothetically,
automotive NK cells have an additional favorable toxic effect in
comparison to CAR T cells, particularly for avoiding unfavorable
effects such as CRS. Additionally, in contrast to T cells, donor NK
cells do not target non-hematopoietic cells, indicating that NK
cell-mediated antitumor activity may be activated in the absence of
graft-vs.-host disease (90)
(Fig. 4).
CRISPR is a gene modifying tool that uses a guide
gRNA to modify a DNA sequence. As this technology is an
integration-free gene incorporation system, it offers a foolproof
and competent gene knock-in process. Following the significant
progress associated with CRISPR technology, it has the potential to
emerge as a promising immunotherapy (91). The CRISPR system can be directly
applied to mammalian cells via transfection using a plasmid that
contains both nuclease and sgRNA. Cas9 encodes a large molecule
with a multifunctional DNA endonuclease and is known to excise
dsDNA from 3-bp upstream of the protospacer adjacent motif (PAM)
(92). Once the nuclease binds to its
gRNA, the compound scans for an integral target DNA sequence
(93). The PAM sequence has a
significant role in recognizing self and non-self sequences. The
local PAM sequence is usually used as a ‘spy’ on the nuclease
sequence 5-NGG-3, in which N is any of the four deoxyribonucleic
acid bases (94).
Recently published reports suggest that CRISPR
technology can deliver the CAR gene to the TRAC locus of T cells.
In this regard, CRISPR-edited universal-CAR T cell therapy has been
used in humans (NCT03166878 and NCT03229876). This technology is
rapidly developing with the potential for gene correction (82). It is reported that anti-CD19 CAR to
the TCR α constant locus (TRAC locus) not only results in increased
T cell potency but also in the consistent expression of CAR in
peripheral blood T cells (95).
CRISPR-modified cells have been shown to perform well in generally
constructed CAR T cells in a mouse model of ALL (96). It has also been demonstrated that
targeting the TRAC locus turns on CAR signaling, thereby initiating
successful internalization and stimulation of the CAR for single
and repeated exposure to a tumor antigen. It also delays effector T
cell differentiation and exhaustion (97). Multiplex genome editing is another
attractive application of the CRISPR/Cas9 tool. Proficient genomic
disturbance of multiple gene loci to create a universal donor cell
and a potent effector T lymphocyte targeted to different inhibitory
pathways, such as PD-1 and CTLA4, is established by incorporating
several gRNAs into a CAR vector. Furthermore, two-fold knockout of
the TCR gene and HLA class I can effectively permit the generation
of an allogeneic uCAR T cell, in addition to CAR T cells that are
universally Fas-resistant via three-fold gene disruption (98).
The most notable advantage of CAR T cell therapy
over other cancer therapies is the abrupt time intervention and
single infusion of CAR T cells. Additionally, 2–3 weeks of proper
care and observation is sufficient for the patient. CAR T cell
therapy is regarded as a ‘drug of the present day’ and its efficacy
may persist for decades as the cells can survive in the host body
in the long term, with a constant ability to find and destroy
cancer cells during relapse (2,3).
Currently, CAR T cell therapy is licensed for use in patients for
whom transplantation has not been curative and who relapse
following transplant. CAR T cell therapy is expected to be a
substitute for different types of transplant (99). Clinical trials on blood cancer have
shown that, even in patients with a refractory condition in which
cancer reverted following several transplants, CAR T cell therapy
was successful in completely eradicating the disease (100). Additionally, with CAR T cells,
patients can live life without the risk of relapse and benefit from
a sanatory treatment, such as stem cell transplantation. Therefore,
CAR T cell therapy can be referred to as a ‘living drug’ (101).
Several clinical trials are currently examining the
use of CAR T cell therapy against solid tumors and other diseases.
Reports suggest that mesothelin-specific CAR mRNA-engineered T
cells can induce antitumor activity in solid malignancies (102,103).
Furthermore, CAR technology has been used in organ transplantation
using two novel HLA-A2-specific CARs, one representing a CD28-CD3d
signaling domain (CAR) and the other missing an intracellular
signaling domain (dCAR). The adoptive transfer of allospecific
regulatory T cells (Tregs) provides a better safeguard from graft
rejection compared with that of polyclonal Tregs (104). CAR comprising the ICOS signaling
domain liaises with the effective antitumor effect on epidermal
growth factor receptor variant III (EGFRvIII)-expressing glioma
(105). The preclinical evaluation
of CAR T cell therapy targeting the tumor antigen 5T4 in ovarian
cancer has been associated with a successful outcome (106). An unexpected evolutionary finding
was reported during the investigation of CAR targeting autoimmune
diseases; therapy lacking autoimmune diseases that purposely target
only the disease-causing cells, chimeric autoantibody receptor
(CAAR), contain pemphigus vulgaris autoantigen, desmoglein (Dsg3),
combined to CD137-CD3d signaling domains. Dsg3 CAAR-T cells exhibit
accurate toxicity against anti-Dsg3-expressing cells (107). Certain solid tumor CAR targets are
undergoing research with varied genetic products arising from gene
mutations (EGFRvIII) (108) or
modified glycosylation patterns (MUC1) (109), and cancer-testis antigen-derived
peptides (MAGE), CAR specifically targets certain overexpressed
antigens in breast cancer, lung cancer and pancreatic cancer, such
as carcinoembryonic antigen (110)
GD2, prostate-specific membrane antigen, HER2/ERBB2, MUC16
(111) and mesothelin or
tumor-affiliated stoma (fibroblast activation protein and vascular
endothelial growth factor receptor) (112).
As per the 2018 records on the total clinical trials
conducted in the immuno-oncology domain, 220 trials involving CAR T
therapy were performed to identify specific targets. Successfully
developed CAR T cell drugs that are available to the market include
CTL019 (Kymriah) (113), KTE-C19
(Yescarta) (114) and JCAR015
(115). These have been developed by
companies known to be antecedents of CAR T cell therapy
development, such as Novartis in association with the University of
Pennsylvania, Kite Pharma with National Cancer Institute, and Juno
Therapeutics with Sloan Kettering, respectively, and are used to
treat ALL, NHL and ALL. These CAR T therapies represent a defining
moment in 2017 in the field of oncology. The first two therapies
specific to CD19 and approved by the FDA included Kymriah
(tisagenlecleucel-T) and Yescarta (axicabtageneciloleucel) by
Novartis and Kite Pharma/Gilead Sciences, respectively (116).
The global ELIANA trial reported a high success
rate, with a 3-month complete remission rate of 83% with
tisagenlecleucel, and a 6-month survival rate of 89% (117). Another trial, ZUMA-1, reported an
equivalent results with an 82% overall response rate and 54% of
complete remission rate following a single infusion of the
therapeutic regime in 8 months. This indicates that the remission
rate was more pronounced in pediatric B-ALL than that in adult
relapsed/refractory DLBCL; however, reactivity was high in both
diseases (118).
Furthermore, multiple clinical investigations have
been implemented by Cellectis following UCART19. Cellectis is
currently leading with two successful FDA Investigational New Drug
(IND)-approved allogeneic CAR T approaches (119): UCART123 for patients with blastic
plasmacytoid dendritic cell neoplasm and AML (120) and UCART22 IND for patients with
B-ALL (121). Two other ongoing
clinical trials are UCARTCS1 for suppressing CS1-expressing
hematologic malignancies (122) and
UCART38 for CD38-expressing hematologic malignancies. UCART38 is
specifically developed to target T cell ALL, multiple myeloma,
mantle cell lymphoma and NHL (123,124). A
list of CAR therapy clinical trials are listed in Table I.
Although immunotherapy has achieved clinical success
in treating blood cancer, the same success rates have not been
observed for solid tumors. The various challenges associated with
its safety, cost-effectiveness and quality require thorough
investigation in order to implement this therapy for all cancer
types. Another approach is to integrate CAR T cells with different
types of immunotherapy to enhance its effectiveness. For example,
CARs may be combined with certain checkpoint inhibitors, which
limit tumor defense mechanisms against T cells. It is expected that
future CAR T cell therapy regimens will target several diverse
molecules for a particular type of tumor, such that CAR T cells can
efficiently recognize cancerous cells even if these undergo
mutations in their target molecules.
A number of CAR T therapies are already available to
the market, but these are expensive, for example, $475,000
(€400,000) for Yescarta and $373,000 (€316,000) for Kymriah.
According to experts, when hospitalization expenses and the costs
of other drugs required for the treatment are also considered, the
cost increases to almost $1,500,000 per cancer patient. Therefore,
a possible solution is to reduce the cost of allogeneic CAR T
treatment by supplying T lymphocytes from a healthy individual that
can be readily utilized when a patient requires it, rather than
genetically modifying each patient's T cells individually.
Considerable scientific challenges also exist with regard to
immunology, in addition to manufacturing, transportation and
banking solutions to enhance the extensive treatment of patients.
Therefore, scientists are investigating measures to overcome
clinical challenges in terms of regulations. CAR T cells are
available in several scientific frameworks, which may vary widely
in different countries. These combined challenges and technology
require standardization; however, CAR T cells offer patients hope
of advanced treatment. As the first therapy is already available in
the market, there is potential for a specific and improved
alternative becoming available in upcoming decades.
We acknowledge the Science and Engineering Research
Board for the generous funding.
Funding from the Science and Engineering Board,
Department of Science and Technology, Government of India is duly
acknowledged. ECR/2016/000584.
Not applicable.
RM and NG conceptualized and co-wrote the
manuscript. CRC, SA, PS and PG contributed to the literature
review, organization and writing of various sections of the
manuscript. NG is the PI and grant holder.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Restifo NP, Dudley ME and Rosenberg SA:
Adoptive immunotherapy for cancer: Harnessing the T cell response.
Nat Rev Immunol. 12:269–281. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Galluzzi L and Martin P: CARs on a highway
with roadblocks. Oncoimmunology. 6:e13884862017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Perales MA, Kebriaei P, Kean LS and
Sadelain M: Building a safer and faster CAR: Seatbelts, airbags,
and CRISPR. Biol Blood Marrow Transplant. 24:27–31. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sharpe M and Mount N: Genetically modified
T cells in cancer therapy: Opportunities and challenges. Dis Model
Mech. 8:337–350. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McGuirk J, Waller EK, Qayed M, Abhyankar
S, Ericson S, Holman P, Keir C and Myers GD: Building blocks for
institutional preparation of CTL019 delivery. Cytotherapy.
19:1015–1024. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prasad V: Immunotherapy:
Tisagenlecleucel-the first approved CAR-T-cell therapy:
Implications for payers and policy makers. Nat Rev Clin Oncol.
15:11–12. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Neelapu SS, Tummala S, Kebriaei P, Wierda
W, Gutierrez C, Locke FL, Komanduri KV, Lin Y, Jain N, Daver N, et
al: Chimeric antigen receptor T-cell therapy-assessment and
management of toxicities. Nat Rev Clin Oncol. 15:47–62. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miliotou AN and Papadopoulou LC: CAR
T-cell therapy: A new era in cancer immunotherapy. Curr Pharm
Biotechnol. 19:5–18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mirzaei HR, Jamali A, Jafarzadeh L,
Masoumi E, Alishah K, Fallah Mehrjardi K, Emami SAH, Noorbakhsh F,
Till BG and Hadjati J: Construction and functional characterization
of a fully human anti-CD19 chimeric antigen receptor
(huCAR)-expressing primary human T cells. J Cell Physiol.
234:9207–9215. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vormittag P, Gunn R, Ghorashian S and
Veraitch FS: A guide to manufacturing CAR T cell therapies. Curr
Opin Biotechnol. 53:164–181. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Abate-Daga D and Davila ML: CAR models:
Next-generation CAR modifications for enhanced T-cell function. Mol
Ther Oncolytics. 3:160142016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang QY, Yang JD and Wang YS: Current
strategies to improve the safety of chimeric antigen receptor (CAR)
modified T cells. Immunol Lett. 190:201–205. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mirzaei HR, Mirzaei H, Namdar A, Rahmati
M, Till BG and Hadjati J: Predictive and therapeutic biomarkers in
chimeric antigen receptor T-cell therapy: A clinical perspective. J
Cell Physiol. 234:5827–5841. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dai H, Wang Y, Lu X and Han W: Chimeric
antigen receptors modified T-cells for cancer therapy. J Natl
Cancer Inst. 108(pii): djv4392016.PubMed/NCBI
|
|
15
|
Mirzaei HR, Mirzaei H, Lee SY, Hadjati J
and Till BG: Prospects for chimeric antigen receptor (CAR) γδ T
cells: A potential game changer for adoptive T cell cancer
immunotherapy. Cancer Lett. 380:413–423. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morgan RA: Chapter 17-Adoptive T-cell
therapy: Engineering T-cell receptors. Cancer Immunother (Sec Ed).
261–272. 2013. View Article : Google Scholar
|
|
17
|
Louis CU, Savoldo B, Dotti G, Pule M, Yvon
E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, et al: Antitumor
activity and long-term fate of chimeric antigen receptor-positive T
cells in patients with neuroblastoma. Blood. 118:6050–6056. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Duong CP, Yong CS, Kershaw MH, Slaney CY
and Darcy PK: Cancer immunotherapy utilizing gene-modified T cells:
From the bench to the clinic. Mol Immunol. 67:46–57. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sadelain M: CAR therapy: The CD19
paradigm. J Clin Invest. 125:3392–3400. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eshhar Z: The T-body approach: Redirecting
T cells with antibody specificity. Handb Exp Pharmacol. 329–342.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brentjens R, Yeh R, Bernal Y, Riviere I
and Sadelain M: Treatment of chronic lymphocytic leukemia with
genetically targeted autologous T cells: Case report of an
unforeseen adverse event in a phase I clinical trial. Mol Ther.
18:666–668. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Till BG, Jensen MC, Wang J, Qian X, Gopal
AK, Maloney DG, Lindgren CG, Lin Y, Pagel JM, Budde LE, et al:
CD20-specific adoptive immunotherapy for lymphoma using a chimeric
antigen receptor with both CD28 and 4-1BB domains: Pilot clinical
trial results. Blood. 119:3940–3950. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Park JH and Brentjens RJ: Are all chimeric
antigen receptors created equal? J Clin Oncol. 33:651–653. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qian L, Li D, Ma L, He T, Qi F, Shen J and
Lu XA: The novel anti-CD19 chimeric antigen receptors with
humanized scFv (single-chain variable fragment) trigger leukemia
cell killing. Cell Immunol. 304-305:49–54. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lock D, Mockel-Tenbrinck N, Drechsel K,
Barth C, Mauer D, Schaser T, Kolbe C, Al Rawashdeh W, Brauner J,
Hardt O, et al: Automated manufacturing of potent CD20-directed
chimeric antigen receptor T cells for clinical use. Hum Gene Ther.
28:914–925. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tang XY, Sun Y, Zhang A, Hu GL, Cao W,
Wang DH, Zhang B and Chen H: Third-generation CD28/4-1BB chimeric
antigen receptor T cells for chemotherapy relapsed or refractory
acute lymphoblastic leukaemia: A non-randomised, open-label phase I
trial protocol. BMJ Open. 6:e0139042016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yeku OO and Brentjens RJ: Armored CAR
T-cells: Utilizing cytokines and pro-inflammatory ligands to
enhance CAR T-cell anti-tumour efficacy. Biochem Soc Trans.
44:412–418. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chmielewski M and Abken H: TRUCKs: The
fourth generation of CARs. Expert Opin Biol Ther. 15:1145–1154.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kueberuwa G, Kalaitsidou M, Cheadle E,
Hawkins RE and Gilham DE: CD19 CAR T cells expressing IL-12
eradicate lymphoma in fully lymphoreplete mice through induction of
host immunity. Mol Ther Oncolytics. 8:41–51. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kerkar SP, Muranski P, Kaiser A, Boni A,
Sanchez-Perez L, Yu Z, Palmer DC, Reger RN, Borman ZA, Zhang L, et
al: Tumor-specific CD8+ T cells expressing interleukin-12 eradicate
established cancers in lymphodepleted hosts. Cancer Res.
70:6725–6734. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Giacca M and Zacchigna S: Virus-mediated
gene delivery for human gene therapy. J Control Release.
161:377–388. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yi Y, Noh MJ and Lee KH: Current Advances
in retroviral gene therapy. Curr Gene Ther. 11:218–228. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kidd ME, Shin S and Shea LD: Fibrin
hydrogels for lentiviral gene delivery in vitro and in vivo. J
Control Release. 157:80–85. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Munñoz-López M and García-Perez JL: DNA
transposons: Nature and applications in genomics. Curr Genomics.
11:115–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Morita D, Nishio N, Saito S, Tanaka M,
Kawashima N, Okuno Y, Suzuki S, Matsuda K, Maeda Y, Wilson MH, et
al: Enhanced expression of Anti-CD19 chimeric antigen receptor in
piggyBac transposon-engineered T cells. Mol Ther Methods Clin Dev.
8:131–140. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kebriaei P, Singh H, Huls MH, Figliola MJ,
Bassett R, Olivares S, Jena B, Dawson MJ, Kumaresan PR, Su S, et
al: Phase I trials using sleeping beauty to generate CD19-specific
CAR T cells. J Clin Invest. 126:3363–3376. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Manuri PV, Wilson MH, Maiti SN, Mi T,
Singh H, Olivares S, Dawson MJ, Huls H, Lee DA, Rao PH, et al:
piggyBac Transposon/Transposase system to generate CD19-specific T
cells for the treatment of B-lineage malignancies. Hum Gene Ther.
21:427–437. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yarmush ML, Golberg A, Serša G, Kotnik T
and Miklavčič D: Electroporation-based technologies for medicine:
Principles, applications, and challenges. Annu Rev Biomed Eng.
16:295–320. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chicaybam L, Sodre AL, Curzio BA and
Bonamino MH: An efficient low cost method for gene transfer to T
lymphocytes. PLoS One. 8:e602982013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chicaybam L, Barcelos C, Peixoto B,
Carneiro M, Limia CG, Redondo P, Lira C, Paraguassú-Braga F,
Vasconcelos ZF, Barros L and Bonamino MH: An efficient
electroporation protocol for the genetic modification of mammalian
cells. Front Bioeng Biotechnol. 4:992017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kotnik T, Frey W, Sack M, Haberl Meglič S,
Peterka M and Miklavčič D: Electroporation-based applications in
biotechnology. Trends Biotechnol. 33:480–488. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ramamoorth M and Narvekar A: Non viral
vectors in gene therapy-an overview. J Clin Diagn Res. 9:GE01–GE06.
2015.PubMed/NCBI
|
|
43
|
Yin H, Kauffman KJ and Anderson DG:
Delivery technologies for genome editing. Nat Rev Drug Discov.
16:387–399. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Smith TT, Stephan SB, Moffett HF, McKnight
LE, Ji W, Reiman D, Bonagofski E, Wohlfahrt ME, Pillai SPS and
Stephan MT: In situ programming of leukaemia-specific T cells using
synthetic DNA nanocarriers. Nat Nanotechnol. 12:813–820. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Miller MA: Nanoparticles improve economic
mileage for CARs. Sci Transl Med. 9(pii): eaan27842017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Smith TT, Moffett HF, Stephan SB, Opel CF,
Dumigan AG, Jiang X, Pillarisetty VG, Pillai SPS, Wittrup KD and
Stephan MT: Biopolymers codelivering engineered T cells and STING
agonists can eliminate heterogeneous tumors. J Clin Invest.
127:2176–2191. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dowaidar M, Abdelhamid HN, Hällbrink M,
Freimann K, Kurrikoff K, Zou X and Langel Ü: Magnetic nanoparticle
assisted Self-assembly of cell penetrating
peptides-oligonucleotides complexes for gene delivery. Sci Rep.
7:91592017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kebriaei P: CAR T-cell therapies:
Overcoming the challenges and new strategies. Clin Lymphoma Myeloma
Leuk. 17 (Suppl 2):S74–S78. 2017. View Article : Google Scholar
|
|
49
|
Bonifant CL, Jackson HJ, Brentjens RJ and
Curran KJ: Toxicity and management in CAR T-cell therapy. Mol Ther
Oncolytics. 3:160112016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Brudno JN and Kochenderfer JN: Toxicities
of chimeric antigen receptor T cells: Recognition and management.
Blood. 127:3321–3330. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Neelapu SS, Locke FL, Bartlett NL, Lekakis
LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T,
Lin Y, et al: Axicabtagene Ciloleucel CAR T-cell therapy in
refractory large B-cell lymphoma. N Engl J Med. 377:2531–2544.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Paietta E: Immunobiology of acute
leukemia. In: Neoplastic diseases of the bloodSpringer; Cham: pp.
237–279. 2018
|
|
53
|
Zah E, Lin MY, Jensen MC, Silva-Benedict A
and Chen YY: Abstract IA12: Combating antigen escape with CD19/CD20
bispecific CAR-T cell therapy. Cancer Immunol Res 5 (3 Suppl).
IA122017.
|
|
54
|
Majzner RG and Mackall CL: Tumor antigen
escape from CAR T-cell therapy. Cancer Discov. 8:1219–1226. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhu M, Wu B, Brandl C, Johnson J, Wolf A,
Chow A and Doshi S: Blinatumomab, a Bispecific T-cell Engager
(BiTE(®)) for CD-19 targeted cancer immunotherapy:
Clinical pharmacology and its implications. Clin Pharmacokinet.
55:1271–1288. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Martyniszyn A, Krahl AC, André MC, Hombach
AA and Abken H: CD20-CD19 Bispecific CAR T cells for the treatment
of B-cell malignancies. Hum Gene Ther. 28:1147–1157. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhu F, Shah N, Xu H, Schneider D, Orentas
R, Dropulic B, Hari P and Keever-Taylor CA: Closed-system
manufacturing of CD19 and dual-targeted CD20/19 chimeric antigen
receptor T cells using the CliniMACS prodigy device at an academic
medical center. Cytotherapy. 20:394–406. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Sun LL, Ellerman D, Mathieu M,
Hristopoulos M, Chen X, Li Y, Yan X, Clark R, Reyes A, Stefanich E,
et al: Anti-CD20/CD3 T cell-dependent bispecific antibody for the
treatment of B cell malignancies. Sci Transl Med. 7:287ra702015.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang W, Liu Y, Wang Y, Wang C, Yang QM,
Zhu HL and Han WD: Long-term safety and efficacy of CART-20 cells
in patients with refractory or relapsed B-cell non-Hodgkin
lymphoma: 5-years follow-up results of the phase I and IIa trials.
Signal Transduct Target Ther. 2:170542017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hegde M, Mukherjee M, Grada Z, Pignata A,
Landi D, Navai SA, Wakefield A, Fousek K, Bielamowicz K, Chow KK,
et al: Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor
antigen escape. J Clin Invest. 126:3036–3052. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Schneider D, Xiong Y, Wu D, Nӧlle V,
Schmitz S, Haso W, Kaiser A, Dropulic B and Orentas RJ: A tandem
CD19/CD20 CAR lentiviral vector drives on-target and off-target
antigen modulation in leukemia cell lines. J Immunother Cancer.
5:422017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Grada Z, Hegde M, Byrd T, Shaffer DR,
Ghazi A, Brawley VS, Corder A, Schönfeld K, Koch J, Dotti G, et al:
TanCAR: A novel bispecific chimeric antigen receptor for cancer
immunotherapy. Mol Ther Nucleic Acids. 2:e1052013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ahmed N, Brawley V, Hegde M, Bielamowicz
K, Kalra M, Landi D, Robertson C, Gray TL, Diouf O, Wakefield A, et
al: HER2-specific chimeric antigen receptor-modified virus-specific
T cells for progressive glioblastoma: A phase 1 dose-escalation
trial. JAMA Oncol. 3:1094–1101. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Bielamowicz K, Fousek K, Byrd TT, Samaha
H, Mukherjee M, Aware N, Wu MF, Orange JS, Sumazin P, Man TK, et
al: Trivalent CAR T cells overcome interpatient antigenic
variability in glioblastoma. Neuro Oncol. 20:506–518. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ott PA, Hodi FS and Robert C: CTLA-4 and
PD-1/PD-L1 blockade: New immunotherapeutic modalities with durable
clinical benefit in melanoma patients. Clin Cancer Res.
19:5300–5309. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Curran MA, Montalvo W, Yagita H and
Allison JP: PD-1 and CTLA-4 combination blockade expands
infiltrating T cells and reduces regulatory T and myeloid cells
within B16 melanoma tumors. Proc Natl Acad Sci USA. 107:4275–4280.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hodi FS, O'Day SJ, McDermott DF, Weber RW,
Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel
JC, et al: Improved Survival with Ipilimumab in Patients with
Metastatic Melanoma. N Engl J Med. 363:711–723. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Iwai Y, Ishida M, Tanaka Y, Okazaki T,
Honjo T and Minato N: Involvement of PD-L1 on tumor cells in the
escape from host immune system and tumor immunotherapy by PD-L1
blockade. Proc Natl Acad Sci USA. 99:12293–12297. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Iwai Y, Hamanishi J, Chamoto K and Honjo
T: Cancer immunotherapies targeting the PD-1 signaling pathway. J
Biomed Sci. 24:262017. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ansell SM, Lesokhin AM, Borrello I,
Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry
D, Freeman GJ, et al: PD-1 Blockade with Nivolumab in relapsed or
refractory Hodgkin's lymphoma. N Engl J Med. 372:311–319. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chen R, Zinzani PL, Fanale MA, Armand P,
Johnson NA, Brice P, Radford J, Ribrag V, Molin D, Vassilakopoulos
TP, et al: Phase II study of the efficacy and safety of
pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J
Clin Oncol. 35:2125–2132. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Barbee MS, Ogunniyi A, Horvat TZ and Dang
TO: Current status and future directions of the immune checkpoint
inhibitors ipilimumab, pembrolizumab, and nivolumab in oncology.
Ann Pharmacother. 49:907–937. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Curran MA, Kim M, Montalvo W, Al-Shamkhani
A and Allison JP: Combination CTLA-4 blockade and 4-1BB activation
enhances tumor rejection by increasing T-cell infiltration,
proliferation, and cytokine production. PLoS One. 6:e194992011.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Seidel JA, Otsuka A and Kabashima K:
Anti-PD-1 and Anti-CTLA-4 therapies in cancer: Mechanisms of
action, efficacy, and limitations. Front Oncol. 8:862018.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ren J, Liu X, Fang C, Jiang S, June CH and
Zhao Y: Multiplex genome editing to generate universal CAR T cells
resistant to PD1 inhibition. Clin Cancer Res. 23:2255–2266. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Azijli K, Stelloo E, Peters GJ and Van Den
Eertwegh AJ: New developments in the treatment of metastatic
melanoma: Immune checkpoint inhibitors and targeted therapies.
Anticancer Res. 34:1493–1505. 2014.PubMed/NCBI
|
|
77
|
Deng L, Liang H, Burnette B, Beckett M,
Darga T, Weichselbaum RR and Fu YX: Irradiation and anti-PD-L1
treatment synergistically promote antitumor immunity in mice. J
Clin Invest. 124:687–695. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Shi H, Sun M, Liu L and Wang Z: Chimeric
antigen receptor for adoptive immunotherapy of cancer: Latest
research and future prospects. Mol Cancer. 13:2192014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Liu L, Sun M and Wang Z: Adoptive T-cell
therapy of B-cell malignancies: Conventional and physiological
chimeric antigen receptors. Cancer Lett. 316:1–5. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Minutolo NG, Hollander EE and Powell DJ
Jr: The emergence of universal immune receptor T cell therapy for
cancer. Front Oncol. 9:1762019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhao J, Lin Q, Song Y and Liu D: Universal
CARs, universal T cells, and universal CAR T cells. J Hematol
Oncol. 11:1322018. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lohmueller JJ, Ham JD, Kvorjak M and Finn
OJ: mSA2 affinity-enhanced biotin-binding CAR T cells for universal
tumor targeting. Oncoimmunology. 7:e13686042018. View Article : Google Scholar
|
|
83
|
Qasim W, Zhan H, Samarasinghe S, Adams S,
Amrolia P, Stafford S, Butler K, Rivat C, Wright G, Somana K, et
al: Molecular remission of infant B-ALL after infusion of universal
TALEN gene-edited CAR T cells. Sci Transl Med. 9:eaaj20132017.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Glienke W, Esser R, Priesner C, Suerth JD,
Schambach A, Wels WS, Grez M, Kloess S, Arseniev L and Koehl U:
Advantages and applications of CAR-expressing natural killer cells.
Front Pharmacol. 6:212015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Klingemann H: Are natural killer cells
superior CAR drivers? Oncoimmunology. 3:e281472014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Jiang H, Zhang W, Shang P, Zhang H, Fu W,
Ye F, Zeng T, Huang H, Zhang X, Sun W, et al: Transfection of
chimeric anti-CD138 gene enhances natural killer cell activation
and killing of multiple myeloma cells. Mol Oncol. 8:297–310. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Boissel L, Betancur-Boissel M, Lu W,
Krause DS, Van Etten RA, Wels WS and Klingemann H: Retargeting
NK-92 cells by means of CD19- and CD20-specific chimeric antigen
receptors compares favorably with antibody-dependent cellular
cytotoxicity. Oncoimmunology. 2:e265272013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Romanski A, Uherek C, Bug G, Seifried E,
Klingemann H, Wels WS, Ottmann OG and Tonn T: CD19-CAR engineered
NK-92 cells are sufficient to overcome NK cell resistance in B-cell
malignancies. J Cell Mol Med. 20:1287–1294. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Hermanson DL and Kaufman DS: Utilizing
chimeric antigen receptors to direct natural killer cell activity.
Front Immunol. 6:1952015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu
T, Yin J, You F, Zhu M, Shen W, et al: First-in-man clinical trial
of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients
with relapsed and refractory acute myeloid leukemia. Am J Cancer
Res. 8:1083–1089. 2018.PubMed/NCBI
|
|
91
|
Hsu PD, Lander ES and Zhang F: Development
and Applications of CRISPR-Cas9 for genome engineering. Cell.
157:1262–1278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Haurwitz RE, Jinek M, Wiedenheft B, Zhou K
and Doudna JA: Sequence- and Structure-specific RNA processing by a
CRISPR endonuclease. Science. 329:1355–1358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Cox DB, Platt RJ and Zhang F: Therapeutic
genome editing: Prospects and challenges. Nat Med. 21:121–131.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Shang W, Wang F, Fan G and Wang H: Key
elements for designing and performing a CRISPR/Cas9-based genetic
screen. J Genet Genomics. 44:439–449. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Mirzaei HR, Pourghadamyari H, Rahmati M,
Mohammadi A, Nahand JS, Rezaei A and Hadjati J: Gene-knocked out
chimeric antigen receptor (CAR) T cells: Tuning up for the next
generation cancer immunotherapy. Cancer Lett. 423:95–104. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Eyquem J, Mansilla-Soto J, Giavridis T,
van der Stegen SJ, Hamieh M, Cunanan KM, Odak A, Gönen M and
Sadelain M: Targeting a CAR to the TRAC locus with CRISPR/Cas9
enhances tumour rejection. Nature. 543:113–117. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Mou H, Kennedy Z, Anderson DG, Yin H and
Xue W: Precision cancer mouse models through genome editing with
CRISPR-Cas9. Genome Med. 7:532015. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
MacLeod DT, Antony J, Martin AJ, Moser RJ,
Hekele A, Wetzel KJ, Brown AE, Triggiano MA, Hux JA, Pham CD, et
al: Integration of a CD19 CAR into the TCR alpha chain locus
streamlines production of allogeneic gene-edited CAR T cells. Mol
Ther. 25:949–961. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Ren J, Zhang X, Liu X, Fang C, Jiang S,
June CH and Zhao Y: A versatile system for rapid multiplex
genome-edited CAR T cell generation. Oncotarget. 8:17002–17011.
2017.PubMed/NCBI
|
|
100
|
Grupp SA, Laetsch TW, Buechner J,
Bittencourt H, Maude SL, Verneris MR, Myers GD, Boyer MW, Rives S,
De Moerloose B, et al: Analysis of a global registration trial of
the efficacy and safety of CTL019 in pediatric and young adults
with relapsed/refractory acute lymphoblastic leukemia (ALL). Blood.
128:2212016.
|
|
101
|
Zhao Z, Chen Y, Francisco NM, Zhang Y and
Wu M: The application of CAR-T cell therapy in hematological
malignancies: Advantages and challenges. Acta Pharm Sin B.
8:539–551. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Beatty GL, Haas AR, Maus MV, Torigian DA,
Soulen MC, Plesa G and Kalos M: Mesothelin-specific chimeric
antigen receptor mRNA-engineered T cells induce antitumor activity
in solid malignancies. Cancer Immunol Res. 2:112–120. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Mirzaei HR, Rodriguez A, Shepphird J,
Brown CE and Badie B: Chimeric antigen receptors T cell therapy in
solid tumor: Challenges and clinical applications. Front Immunol.
8:18502017. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Boardman DA, Philippeos C, Fruhwirth GO,
Ibrahim MA, Hannen RF, Cooper D, Marelli-Berg FM, Watt FM, Lechler
RI, Maher J, et al: Expression of a chimeric antigen receptor
specific for donor HLA class I enhances the potency of human
regulatory T cells in preventing human skin transplant rejection.
Am J Transplant. 17:931–943. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Shen CJ, Yang YX, Han EQ, Cao N, Wang YF,
Wang Y, Zhao YY, Zhao LM, Cui J, Gupta P, et al: Chimeric antigen
receptor containing ICOS signaling domain mediates specific and
efficient antitumor effect of T cells against EGFRvIII expressing
glioma. J Hematol Oncol. 6:332013. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Owens GL, Sheard VE, Kalaitsidou M, Blount
D, Lad Y, Cheadle EJ, Edmondson RJ, Kooner G, Gilham DE and Harrop
R: Preclinical assessment of CAR T-cell therapy targeting the tumor
antigen 5T4 in ovarian cancer. J Immunother. 41:130–140. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ellebrecht CT, Bhoj VG, Nace A, Choi EJ,
Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis
G, et al: Reengineering chimeric antigen receptor T cells for
targeted therapy of autoimmune disease. Science. 353:179–184. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
O'Rourke DM, Nasrallah MP, Desai A,
Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem
S, Maloney E, Shen A, et al: A single dose of peripherally infused
EGFRvIII-directed CAR T cells mediates antigen loss and induces
adaptive resistance in patients with recurrent glioblastoma. Sci
Transl Med. 9(pii): eaaa09842017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Posey AD, Schwab RD, Boesteanu AC,
Steentoft C, Mandel U, Engels B, Stone JD, Madsen TD, Schreiber K,
Haines KM, et al: Engineered CAR T cells targeting the
cancer-associated Tn-Glycoform of the membrane mucin MUC1 control
adenocarcinoma. Immunity. 44:1444–1454. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Burga RA, Thorn M, Point GR, Guha P,
Nguyen CT, Licata LA, DeMatteo RP, Ayala A, Joseph Espat N,
Junghans RP and Katz SC: Liver myeloid-derived suppressor cells
expand in response to liver metastases in mice and inhibit the
anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother.
64:817–829. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Ahmed N, Brawley VS, Hegde M, Robertson C,
Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, et al:
Human epidermal growth factor receptor 2 (HER2)-specific chimeric
antigen receptor-modified T cells for the immunotherapy of
HER2-positive sarcoma. J Clin Oncol. 33:1688–1696. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Akce M, Zaidi MY, Waller EK, El-Rayes BF
and Lesinski GB: The potential of CAR T cell therapy in pancreatic
cancer. Front Immunol. 9:21662018. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Bach PB, Giralt SA and Saltz LB: FDA
approval of tisagenlecleucel: Promise and complexities of a $475
000 cancer drug. JAMA. 318:1861–1862. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Fala L: Yescarta (Axicabtagene Ciloleucel)
second CAR T-cell therapy approved for patients with certain types
of large B-cell lymphoma. Am Health Drug Benefits. 11:109–111.
2018.
|
|
115
|
Hey SP and Kesselheim AS: The FDA, Juno
therapeutics, and the ethical imperative of transparency. BMJ.
354:i44352016. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Rotolo A, Caputo V and Karadimitris A: The
prospects and promise of chimeric antigen receptor immunotherapy in
multiple myeloma. Br J Haematol. 173:350–364. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Almond LM, Charalampakis M, Ford SJ,
Gourevitch D and Desai A: Myeloid sarcoma: Presentation, diagnosis,
and treatment. Clin Lymphoma Myeloma Leuk. 17:263–267. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Locke FL, Neelapu SS, Bartlett NL, Siddiqi
T, Chavez JC, Hosing CM, Ghobadi A, Budde LE, Bot A, Rossi JM, et
al: Phase 1 results of ZUMA-1: A multicenter study of KTE-C19
Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol
Ther. 25:285–295. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Qasim W, Ciocarlie O, Adams S, Inglott S,
Murphy C, Rivat C, Wright G, Lucchini G, Silva J, Rao K, et al:
Preliminary results of UCART19, an allogeneic anti-CD19 CAR T-cell
product in a first-in-human trial (PALL) in pediatric patients with
CD19+ relapsed/refractory B-cell acute lymphoblastic leukemia.
Blood. 130:12712017.
|
|
120
|
Cai T, Galetto R, Gouble A, Smith J,
Cavazos A, Konoplev S, Lane AA, Guzman ML, Kantarjian HM, Pemmaraju
N, et al: Pre-clinical studies of Anti-CD123 CAR-T cells for the
treatment of blastic plasmacytoid dendritic cell neoplasm (BPDCN).
Blood. 128:40392016.
|
|
121
|
Gouble A, Schiffer-Mannioui C, Thomas S,
Gautron AS, Poirot L and Smith J: Abstract 3763: UCART22: Allogenic
adoptive immunotherapy of leukemia by targeting CD22 with CAR
T-cells. Cancer Res. 77:3763. 2017.
|
|
122
|
Galetto R, Chion-Sotinel I, Gouble A and
Smith J: Allogenic TCRa/CS1 double knockout T-cells bearing an
anti-CS1 chimeric antigen receptor: An improved immunotherapy
approach for the treatment of multiple myeloma. Cancer Res. 76 (14
Suppl):Abstract nr 2289. 2016.
|
|
123
|
Drent E, Groen RW, Noort WA, Themeli M,
Lammerts van Bueren JJ, Parren PW, Kuball J, Sebestyen Z, Yuan H,
de Bruijn J, et al: Pre-clinical evaluation of CD38 chimeric
antigen receptor engineered T cells for the treatment of multiple
myeloma. Haematologica. 101:616–625. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Coulter I: Compositions. US Patent
2018/0228866A1. Filed August 12 2016; issued August 16 2018.
|