Introduction
Pancreatic cancer is the fourth most common cause of
cancer-related mortality in Japan, and patients with pancreatic
cancer have a poor prognosis with an overall 5-year survival rate
of <5% (1). Factors contributing
to the high mortality rates are late diagnosis due to the lack of
early symptoms, a high resistance to treatment and a high invasive
potential, which render the tumor surgically incurable. Due to
resistance to chemotherapy, radiotherapy and immunotherapy, the
radical resection of pancreatic cancer is the only available method
that has the potential to cure the disease. Therefore, the
inhibition of local recurrence and distant metastasis following
surgical resection is crucial.
Longstanding type 2 diabetes is a risk factor for
the development of pancreatic cancer due to insulin resistance and
associated hyperinsulinemia, hyperglycemia and inflammation are
related to the development of cancer (2). Metformin is widely used in the
treatment of type 2 diabetes mellitus and has been investigated due
to its anti-tumor effects (3). In
various types of cancer, metformin has been suggested to exhibit
anti-tumor activities and to prevent cancer development (4–6).
Previous studies have also suggested that there is a positive
association between metformin and overall survival in pancreatic
cancer treatment (7,8). Wan et al reported in a
meta-analysis of data from 36,791 patients with pancreatic cancer,
that metformin treatment was significantly associated with a
favorable overall survival. Subgroup analyses revealed a marked
reduction in the mortality risk of patients with stage I–II disease
treated with metformin and in patients receiving surgery following
treatment with metformin (7). To
date, a number of in vitro and in vivo studies have
been performed to investigate the anti-tumor activity of metformin
(3); however, there are limited
studies available to date describing the effects of metformin on
epithelial-mesenchymal transition (EMT) and metastasis.
EMT is one of the earliest and most crucial steps in
cancer invasion and metastasis (9–12). The
EMT phenotype is characterized by the loss of cell-cell adhesion
and apical-basolateral polarity, and a phenotypic change in which
cells shift from an epithelial morphology to an elongated
fibroblast-like morphology with invasive properties. EMT involves
the upregulation of mesenchymal markers, such as vimentin and
α-smooth muscle actin (αSMA), and the downregulation of epithelial
adhesion molecules, such as E-cadherin and cytokeratins. EMT is
triggered by the interplay of extracellular signals (such as
collagen) and a number of secreted soluble factors, such as
transforming growth factor β1 (TGF-β1), fibroblast growth factor,
epidermal growth factor and hepatocyte growth factor. Among these,
TGF-β has been identified as the main factor involved in EMT in the
tumor microenvironment. TGF-β1 has been shown to activate various
downstream pathways, including Smads, Akt/mammalian target of
rapamycin (mTOR) and mitogen-activated protein kinase (MAPK),
thereby inducing EMT (9).
Recent studies have suggested that metformin
inhibits EMT in several types of cancer (13–19).
However, the effects of metformin on EMT in pancreatic cancer
remain unclear. Hence, the objective of the present study was to
clarify whether metformin inhibits the EMT and liver metastasis of
pancreatic cancer cells.
Materials and methods
Cell lines and culture
The human pancreatic cell lines, PANC-1 and
MIAPaCa-2, were purchased from RIKEN Bioresources Center Cell Bank,
BxPC-3 cells were from DS Pharma Biochemical Co., and the mouse
pancreatic cancer cell line Panc02 was obtained from the National
Cancer Institute. PANC-1, BxPC-3 and Panc02 cells were cultured in
Roswell Park Memorial Institute (RPMI)-1640 medium supplemented
with 10% fetal bovine serum (FBS), L-glutamine and penicillin (100
U/ml)/streptomycin (100 µg/ml). MIAPaCa-2 cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) with low glucose
supplemented with 10% fetal bovine serum (FBS), L-glutamine and
penicillin (100 U/ml)/streptomycin (100 µg/ml). All cell cultures
were maintained at 37°C in a humidified atmosphere containing 5%
CO2.
TGF-β1-induced EMT and metformin
treatment
After the PANC-1, MIAPaCa-2 and BxPC-3 cells were
grown to >80% confluency, they were transferred to a serum-free
culture medium with 10 ng/ml TGF-β1 in a humidified 5%
CO2 atmosphere at 37°C for the induction of EMT. For
metformin treatment, the cells were incubated with 10 mM metformin
at 37°C for 48 h (Wako Pure Chemical Industries, Ltd.) prior to
stimulation with TGF-β1.
Evaluation of cell viability by WST-8
assay
The cells were seeded into 96-well plates at a
density of 5×103 cells/well. The following day, the
cells were treated with or without metformin and incubated at 37°C
up to 48 h. Cell viability was examined by the addition of 10 µl of
5 mg/ml tetrazolium salt solution to the medium of each well and
incubated at 37°C for 4 h, and the absorbance was read at 450 nm
with a microplate reader (SpectraMax M2; Molecular Devices, LLC).
The absolute values of the absorbance were converted to surviving
fraction data and reported as the percentage of living cells
relative to the control.
Evaluation of cell migration by wound
healing assay
The wound-healing assay was performed as previously
described with minor modifications (20,21).
Briefly, cells were grown to 100% confluency in P60 culture dishes,
and starved in serum-free culture medium for 24 h before
scratching. A circle the cell layer of the monolayer approximately
500 µm in diameter was then made by scrapping with a 10-µl
extra-long micro-pipette tip. The cells were washed twice with PBS
and incubated with serum-free culture medium with or without TGF-β1
and metformin. Microphotographs were acquired with a digital camera
at 0, 12 and 24 h after scratching, and the cell-free area was
measured using ImageJ software (version 1.51). The migration area
was evaluated as the cell-free area at 12 and 24 h, as a percentage
of the scratch at 0 h.
Immunocytochemistry
After the PANC-1, MIAPaCA-2, and BxPC-3 cells were
grown to >80% confluency in 35-mm µ-dishes (Ibidi), they were
washed with PBS and fixed with 4% paraformaldehyde for 20 min at
25°C. The cells were then incubated with mouse anti-human-vimentin
(Santa Cruz Biotechnology, Inc.) for 2 h at 25°C followed by 1 h of
incubation with anti-rabbit IgG conjugated with Alexa Fluor 594
(Alexa Fluor 594, Life Technologies; Thermo Fisher Scientific,
Inc.) at 25°C. The cells were observed using an epi-illumination on
a laser scanning confocal microscope (Olympus Corp.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR was performed to detect the mRNA expression
levels of E-cadherin, vimentin and α-SMA. Total RNA was extracted
from cells using the acid guanidinium phenol chloroform method with
Isogen (Nippon Gene Co. Ltd.) from the cultured pancreatic cancer
cells (PANC-1, MIAPaCa-2 and BxPC-3). The isolated RNA was stored
at −80°C until use in RT-qPCR. Subsequently, complementary DNA
(cDNA) was synthesized from reverse-transcribed 1 µg extracted RNA
using the High Capacity cDNA Reverse Transcription kit (Applied
Biosystems). The expression levels of E-cadherin, vimentin, Snail,
ZEB-1 and GAPDH genes were analyzed by the 7300 Real-Time PCR
system (Applied Biosystems) using the DNA-binding dye SYBR-Green to
detect the PCR products. The relative quantification of gene
expression with the RT-qPCR data was performed using the standard
curve method, with GAPDH as a reference gene. The PCR conditions
were as follows: Denaturation at 95°C for 15 sec, primer annealing
and elongation at 60°C for 1 min, followed by melting curve
analysis, in which the temperature was increased from 60 to 95°C.
The sequences of primers used for RT-qPCR were as follows:
E-cadherin sense, 5′-GAAGGTGACAGAGCCTCTGGAT-3′ and antisense,
5′-CATTCCCGTTGGATGACACA-3′; vimentin sense,
5′-ACACCCTGCAATCTTTCAGACA-3′ and antisense,
5′-GATTCCACTTTGCGTTCAAGGT-3′; α-SMA sense,
5′-GACCGAATGCAGAAGGAGAT-3′ and antisense,
5′-CCACCGATCCAGACAGAGTA-3′; GAPDH sense,
5′-ACCACAGTCCATGCCATCACT-3′ and antisense,
5′-CCATCACGCCACAGTTTCC-3′.
Western blot analysis
Cell debris were removed by washing with ice-cold
PBS; the cells were lysed in Lysis Buffer (CelLytic M;
Sigma-Aldrich; Merck KGaA), scraped and incubated on ice for 15
min. Tissues were frozen using liquid nitrogen, crushed, lysed in
lysis buffer, scraped and incubated on ice for 15 min. The
supernatants were collected, and total protein was mixed with an
SDS sample buffer. The Bradford method was used to measure the
protein content; 20 µg protein were used per lane. The protein
lysates were then separated by electrophoresis on 10% SDS-PAGE and
transblotted to a polyvinylidene fluoride membrane (Atto
Corporation). The membrane was blocked with 10% EzBlock (Atto
Corporation) in TBS-T [10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.1%
Tween-20 V/V] for 60 min at room temperature, washed with TBS-T 3
times, and incubated overnight at 4°C with goat anti-mouse-β-actin
(ab8229, Abcam), mouse anti-human-E-cadherin (mab1838, R&D
Systems, Inc.), mouse anti-mouse-α-SMA (a2547, Sigma-Aldrich; Merck
KGaA), mouse anti-human-vimentin (sc32322, Santa Cruz
Biotechnology, Inc.), mouse anti-human-Smad2/3 (sc8332, Santa Cruz
Biotechnology, Inc.), rabbit anti-human-Snail (3879, Cell Signaling
Technology, Inc.), rabbit anti-mouse-phospho-Akt (Ser473) (4058,
Cell Signaling Technology, Inc.), rabbit anti-mouse-Akt (4691, Cell
Signaling Technology, Inc.), rabbit anti-human-phospho-mTOR (2971,
Cell Signaling Technology, Inc.), rabbit anti-human-mTOR (Cell
Signaling Technology, Inc. 2983) and rabbit
anti-human-phospho-Smad2/3 (8828, Cell Signaling Technology, Inc.)
antibodies in TBS-T (diluted 1:500-1:1,000). The membrane was then
washed with TBS-T 3 times and incubated with the secondary
anti-rabbit (616520, GE Healthcare Japan Corporation), anti-goat
(628420, GE Healthcare Japan Corporation) and anti-mouse (054220,
GE Healthcare Japan Corporation) IgG antibodies in TBS-T (diluted
1:5,000-1:10,000) for 1 h at room temperature. Immuno-reactive
proteins were detected using an ECL-kit (ECL plus, GE Healthcare
Bio-Sciences K.K.). The blots were analyzed using ImageJ (version
1.51) software.
Animal models
A total of 33 of C57BL/6 immunocompetent female mice
(aged 6–8 weeks; body weight, 15–17 g) were provided by Shimizu.
Mice were kept at 18–24°C and 40–70% relative humidity, with a 12-h
light/dark cycle. They were provided with free access to water and
food (CE-2; CLEA Japan). Cultured Panc02 cells were collected and
washed twice with PBS. Panc02 cells (3×106) were
injected subcutaneously into both flanks of 3 mice within 30 min of
collection. After 3 weeks, subcutaneous tumors growing to a maximum
diameter of approximately 10 mm were collected as tumor fragments
for subsequent surgical orthotopic implantation (SOI). SOI of tumor
fragments was performed in 30 of the C57/BL6 mice, as previously
described (22). For implantation,
a small 6–10 mm transverse incision was created on the left flank
of the mouse. The tail of the pancreas and spleen was carefully
exposed through this incision, and a single tumor fragment (3
mm3), harvested from a subcutaneous tumor grown on a
mouse, was sewn to the tail of the pancreas using 7-0 nylon
surgical sutures (ELP). The pancreatic tail with the tumor fragment
and spleen were stored in the abdomen, and the incision was sutured
using 4-0 nylon surgical sutures (ELP). Mice were anesthetized with
5% isoflurane during SOI. A total of 15 mice each were used in the
control and metformin groups. Mice were randomly assigned to
receive either daily peritoneal injections of metformin (125 mg/kg)
or normal saline for 28 days. The mice were monitored daily for any
signs of toxicity or abnormalities, including their appetite and
behavior. Body weight for each animal was measured once a week.
Humane euthanasia was performed when mice reached an experimental
endpoint or if their ascites increased and they gained >20% of
their body weight, or if they were very weak and lost >20% of
their body weight. Following SOI, it was difficult to monitor
internal tumor growth exactly until sacrifice. Therefore, body
weight was used as a humane endpoint index for internal tumor
development. At the time of sacrifice, mice were sacrificed by an
intraperitoneal injection of pentobarbital sodium overdose (120
mg/kg). All animal experiments were approved by the Institutional
Animal Care and Use Committee of Kyoto Prefectural University of
medicine under Assurance Number M30-618.
Tumor growth and metastatic pattern
analysis
The animals were sacrificed 28 days following SOI.
Tumor length, width, height and weight were measured and tumor
volume was calculated as follows: Tumor volume = (length × width ×
height)/2. For the evaluation of metastasis, liver and lung
surfaces were observed, and were further pathologically sectioned
and metastases were observed under a microscope. The
paraffin-embedded liver and lung tissues were sectioned at 4-µm
thickness, then hematoxylin and eosin staining was and performed.
Briefly, the sections were deparaffinized and rehydrated at 37°C
with xylene 3 times for 5 min; then with 100% ethanol 2 times for
10 min each; and finally, in a series of 95, 80 and 70% ethanol for
5 min each. The sections were washed with deionized water for 5
min, and stained with hematoxylin for 5 min at room temperature.
The sections were then rinsed with deionized water for 20 min and
stained with eosin for 5 min at room temperature. Gradient
dehydration was performed with 70, 90, 95 and 100% ethanol for 2,
2, 2 and 10 min, respectively, then with xylene 3 times for 5 min
each at room temperature. The sections were sealed with neutral gum
and placed in a ventilated room at room temperature overnight.
Furthermore, western blot analysis, RT-qPCR and
immunohistochemistry were performed for EMT-related markers of the
primary tumors in the pancreas.
Immunohistochemistry of vimentin in
the primary pancreatic tumors
The paraffin-embedded tumor tissues were sectioned
at 4-µm thickness using a microtome cryostat and mounted on
MAS-coated slides. Hemo-De was used to clear the sections 3 times
for 5 min. The sections were incubated with 100% ethanol for 5 min
3 times, 95% ethanol for 5 min, 90% ethanol for 5 min and 70%
ethanol for 5 min to block endogenous peroxidase activity, followed
by rinsing with distilled water for 5 min. The slides were
submerged in DAKO REAL Target Retrieval solution and then heated in
a water bath for 20 min at 95°C for antigen retrieval. After
cooling down to room temperature, they were rinsed with distilled
water for 5 min; Dako Cytomation protein block (Dako, Tokyo, Japan)
was used for 30 min at room temperature to block nonspecific
background. The sections were then incubated overnight at 4°C with
specific primary antibody against vimentin (sc32322, Santa Cruz
Biotechnology, Inc.) diluted at 1:200 with antibody dilution (Dako;
Agilent Technologies, Inc.). After washing the sections 3 times in
PBS Tween-20 for 7 min, they were incubated with secondary antibody
[Histofine Simple Stain mouse MAX PO (rabbit); Nichirei
Biosciences, Inc.] for 30 min at room temperature. After removing
unbound antibodies by washing 3 times in PBS for 7 min,
diaminobenzidine, a chromogen substrate reagent, was used to
visualize the bound antibodies. After counterstaining with Mayer's
hematoxylin, the sections were washed for 30 min, dehydrated in
graded ethanol (70% ethanol for 5 min, 90% ethanol for 5 min, 95%
ethanol for 5 min and 100% ethanol for 5 min 3 times), cleared in
Hemo-De (3 times for 5 min each) and cover-slipped.
Statistical analysis
All analyses were performed using the JMP ver13.0
(SAS Institute, Inc.). The differences between 2 groups were
analyzed with the unpaired Student's t-test and a Chi-square test.
Differences between >2 groups were determined by one-way ANOVA
followed by Tukey's multiple comparison test to compare the mean
values. All data are presented as the means ± standard deviation.
P<0.05 was considered to indicate a statistically significant
difference.
Results
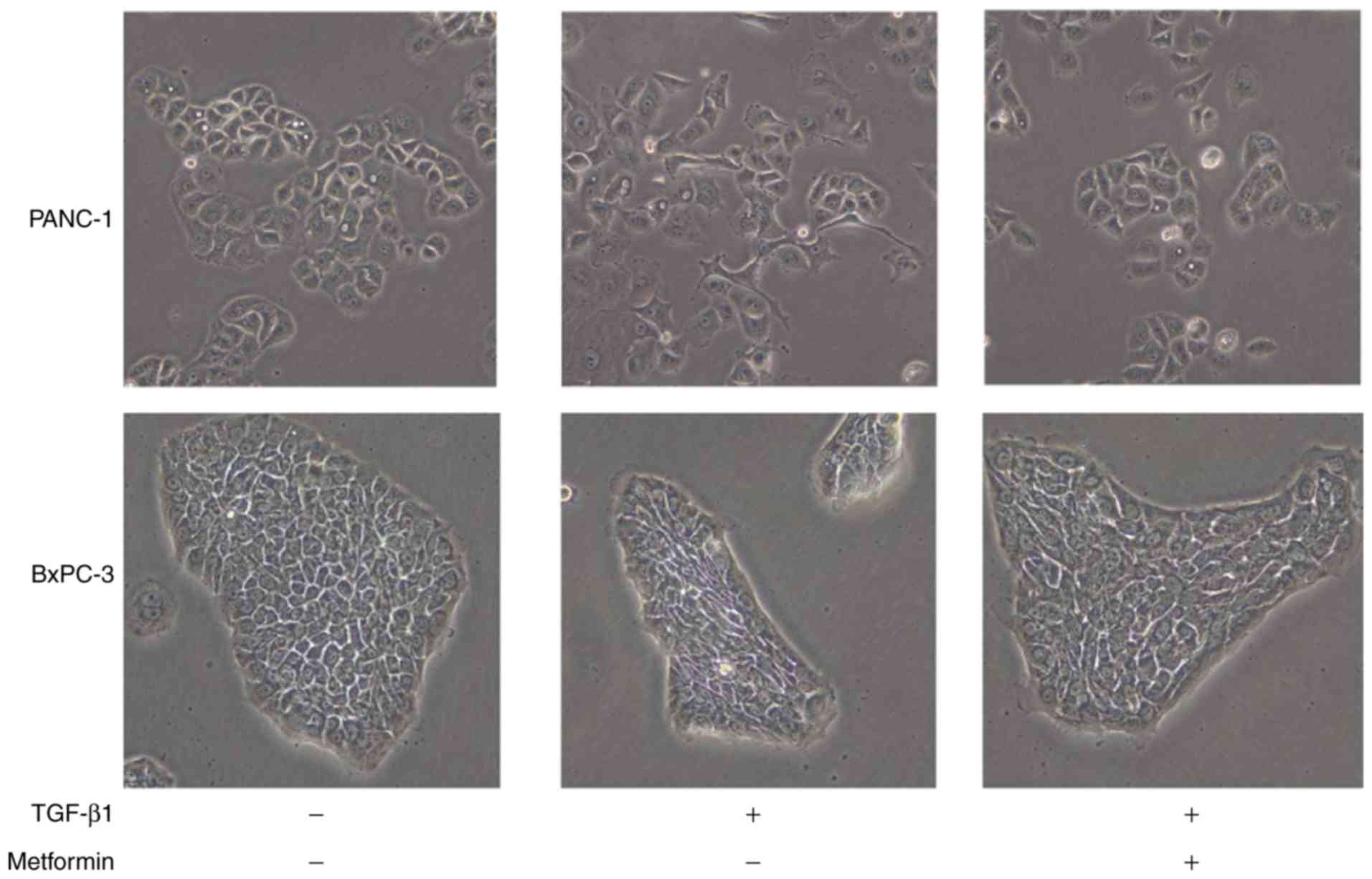
Metformin inhibits morphological
changes consistent with EMT in the presence of TGF-β1
Cell was examined using the WST-8 assay. Cell
viability was not significantly altered by treatment with 10 mM
metformin compared to that in the untreated group (data not shown).
Therefore, the concentration of 10 mM metformin was used for the
subsequent experiments. Following the addition of 10 ng/ml TGF-β1
for 48 h, the PANC-1 and BxPC-3 pancreatic cancer cells acquired an
elongated and fusiform morphology, suggesting that 10 ng/ml TGF-β1
was sufficient to induce EMT. Metformin treatment at 1 h prior to
TGF-β1 exposure inhibited these morphological changes (Fig. 1). The MIAPaCA-2 cell line also an
acquired elongated morphology after TGF-β1 exposure, and metformin
treatment suppressed these morphological changes. However, the
morphological changes of this cell line were smaller than other
cell lines (data not shown).
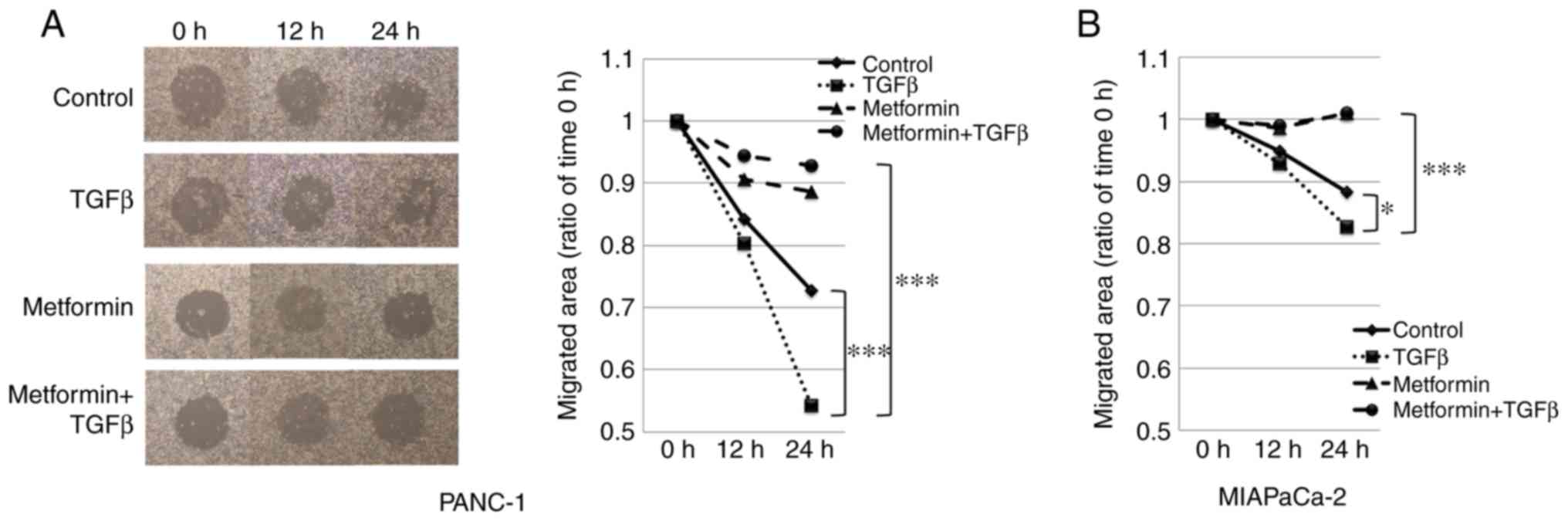
Metformin suppresses the migratory
potential of PANC-1 and MIAPaCa-2 cells
To examine the effect of metformin treatment on the
migratory capability of PANC-1 and MIAPaCa-2 cells, wound healing
assays were performed; at 24 h after scratching, cell migration
into the wound was visualized using a light microscope and images
were captured. The PANC-1 cells treated with TGF-β1 migrated
significantly more rapidly than the control cells after 24 h.
Metformin reduced wound healing in cells treated with TGF-β1 during
the same period (Fig. 2A). Similar
results were observed in the MIAPaCa-2 cells. However, the
TGF-β1-induced migration of MIAPaCa-2 cells was weaker than in that
of the PANC-1 cells. As a result, the difference in the migrated
area between the TGF-β1-treated group and the metformin +
TGF-β1-treated group in the MIAPaCa-2 cells was smaller than that
for the PANC-1 cells; thus, the cell images for the MIAPaCa-2 are
not shown (Fig. 2B). Wounds could
not be made accurately in the BxPC-3 cells due to their strong
adhesion to the plate, and therefore the wound healing assay in
these cells could not be evaluated.
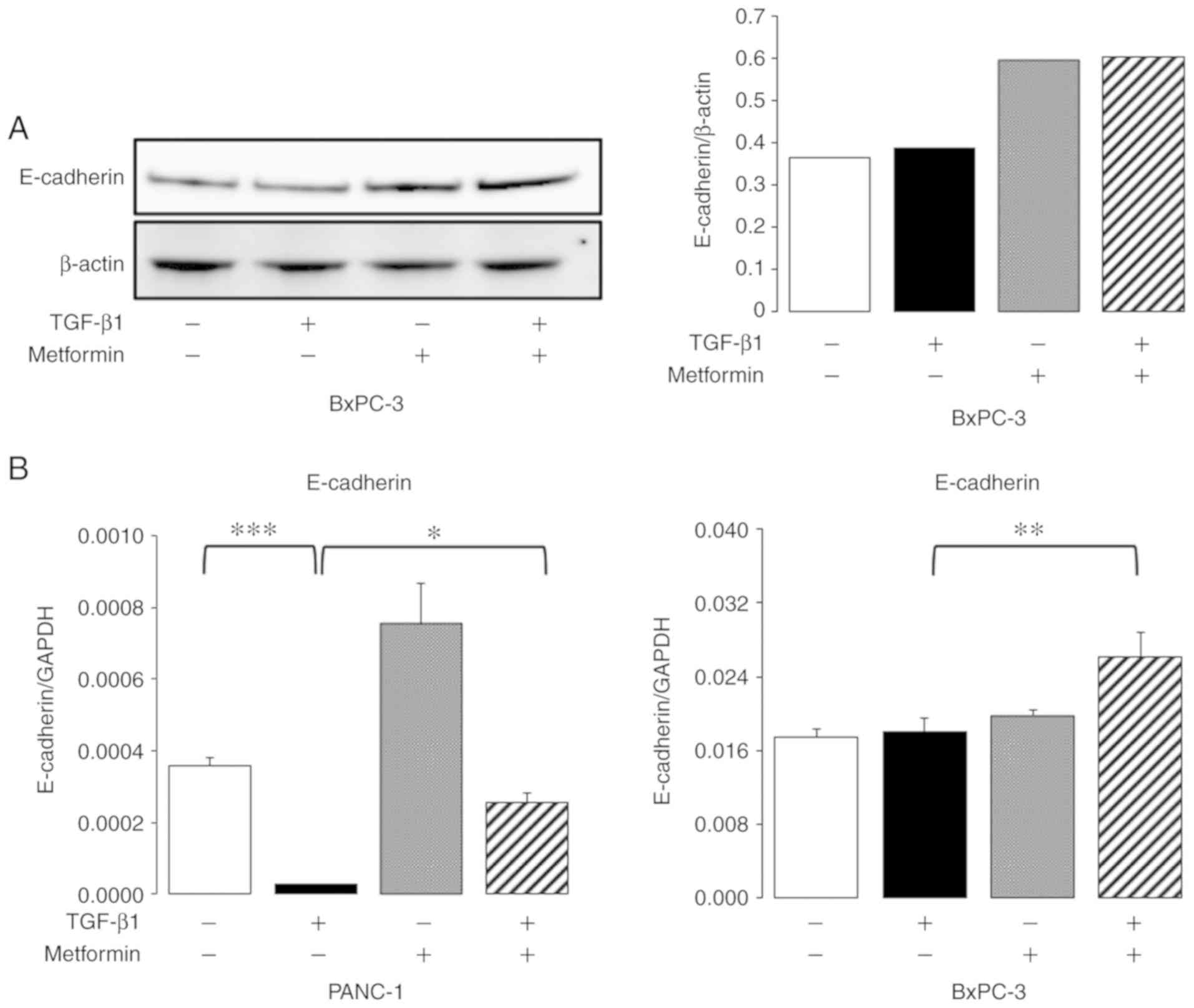
Effect of metformin on molecular
markers of EMT
Western blot analysis and RT-qPCR for E-cadherin
were performed to determine the effects of metformin on EMT. The
results of western blot analysis revealed no marked changes in the
expression of E-cadherin following exposure to TGF-β1, and an
increase in E-cadherin expression was observed following metformin
treatment in the BxPC3 cells (Fig.
3A). However, E-cadherin protein expression was not observed
even in the absence of TGF-β1 in either the PANC-1 or MIAPaCa-2
cells (data not shown). RT-qPCR analysis revealed a decrease in the
mRNA expression of E-cadherin following TGF-β1 exposure, which was
blocked by metformin treatment in the PANC-1 cells (Fig. 3B). The mRNA expression of E-cadherin
was not downregulated by TGF-β1 exposure in the BxPC3 cells, but
was upregulated with a combination of TGF-β1 and metformin
treatment (Fig. 3B).
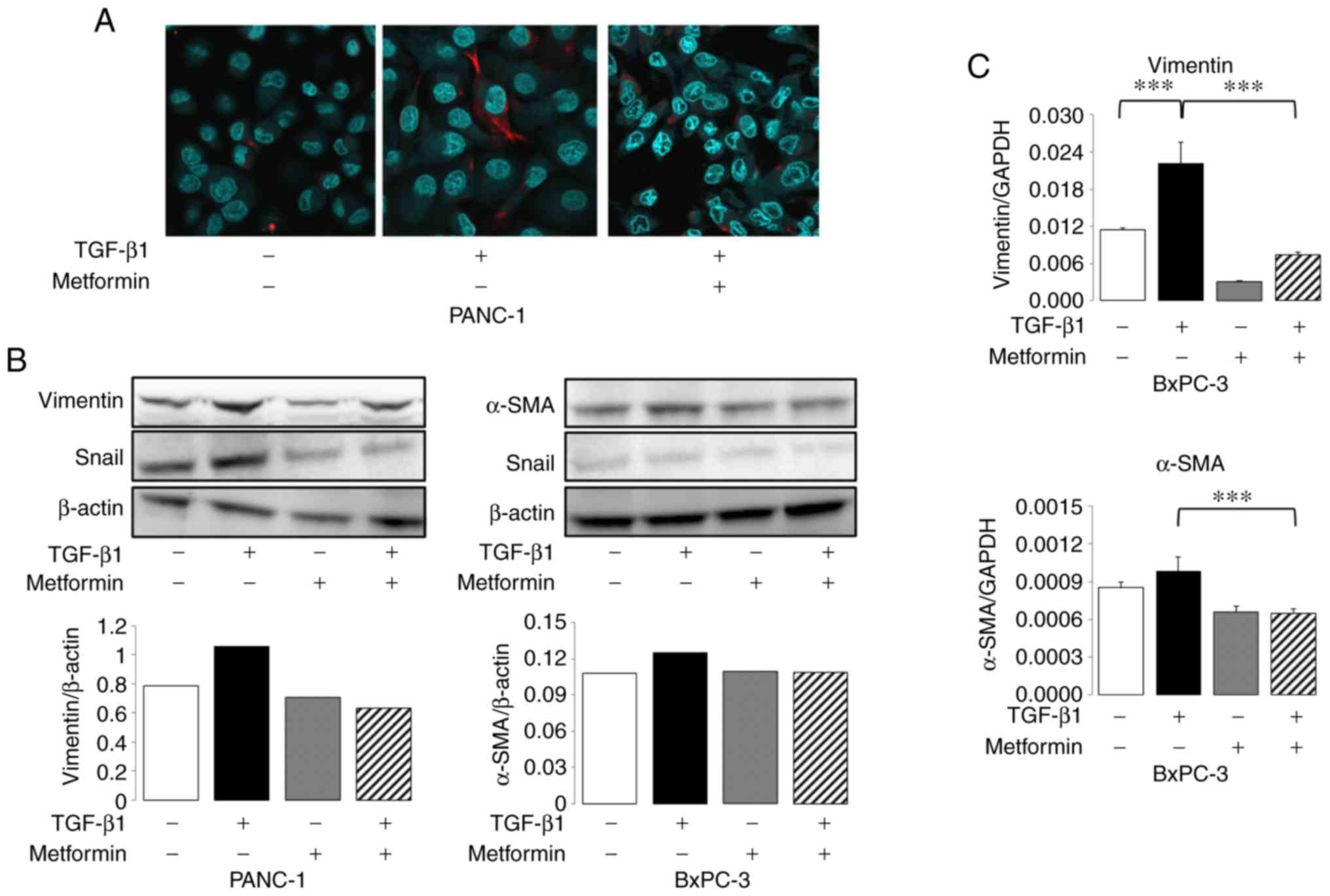
Subsequently, immunocytochemistry, western blot
analysis and RT-qPCR of mesenchymal markers (i.e., vimentin and
α-SMA) and the EMT transcription factor, Snail, were performed.
Immunofluorescence staining for vimentin revealed a strong
expression following exposure to TGF-β1 for 48 h, and a stronger
expression was observed at the tip of spindle-shaped cells, which
was reversed by metformin treatment in the PANC-1 cells (Fig. 4A). Vimentin protein expression was
observed only in PANC-1 cells using immunocytochemistry. As shown
by western blot analysis, the expression of vimentin and Snail was
significantly upregulated by TGF-β1 treatment, which was blocked by
metformin treatment in the PANC-1 cells (Fig. 4B). In the BxPC-3 cells, vimentin
expression was not observed (data not shown); however, the
expression of α-SMA was upregulated by TGF-β1 treatment, which was
blocked by metformin treatment. Furthermore, Snail expression was
downregulated by the combination of TGF-β1 and metformin treatment
(Fig. 4B). The mRNA expression of
vimentin was significantly upregulated by TGF-β1 treatment, which
was blocked by metformin treatment (Fig. 4C). Exposure to TGF-β1 slightly
increased the expression of αSMA, although the change was not
statistically significant, and this slight increase in expression
was blocked by metformin treatment in BxPC-3 cells (Fig. 4C).
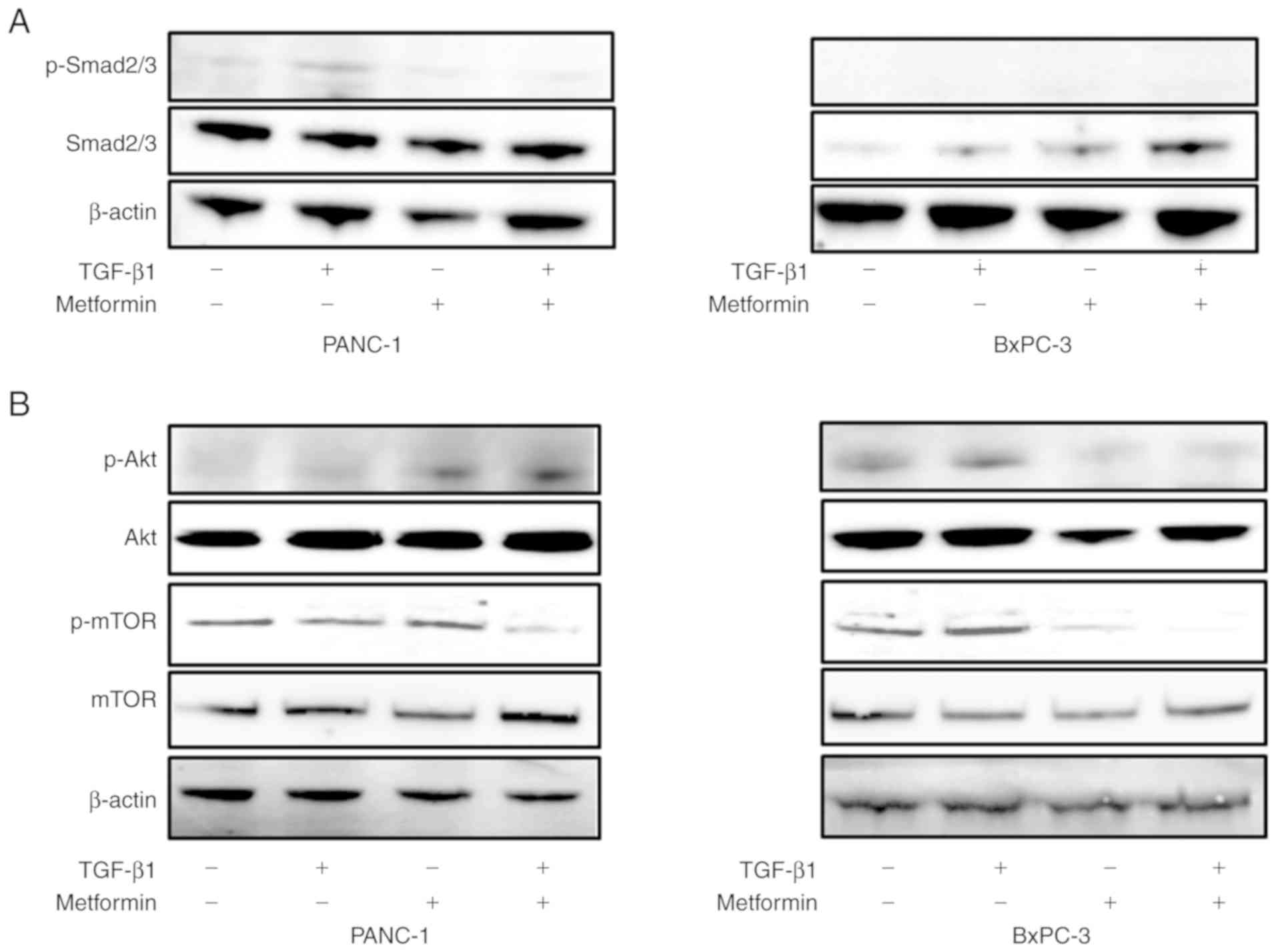
Effect of metformin on the Smad and
Akt/mTOR pathways
To elucidate the mechanisms through which metformin
inhibits TGF-β1-induced EMT, the levels of phosphorylation of
Smad2/3, Akt, and mTOR were examined in the pancreatic cell lines.
First, the role of metformin in the regulation of Smad2/3
expression and phosphorylation, which is involved in the downstream
signaling pathway of TGF-β1, was investigated by western blot
analysis. In the PANC-1 cells, exposure to TGF-β1 resulted in the
phosphorylation of Smad2/3, which was blocked by metformin
treatment (Fig. 5A). However, the
exposure of BxPC-3 cells, which have a naturally low expression of
Smad2/3, to TGF-β1 and metformin did not alter the phosphorylation
of Smad2/3 (Fig. 5A). Subsequently,
other potential pathways of TGF-β1-induced EMT were investigated.
The role of metformin in regulating Akt and mTOR expression and
phosphorylation is involved in the downstream signaling pathway of
TGF-β1. The exposure of the PANC-1 cells to TGF-β1 did not result
in the phosphorylation of Akt and mTOR, suggesting that this
pathway is not involved in TGF-β1-induced EMT in these cells. On
the other hand, in the BxPC-3 cells, exposure to TGF-β1 resulted in
the phosphorylation of Akt and mTOR. Metformin treatment decreased
the expression of Akt. Moreover, in the BxPC cells, metformin
treatment decreased the phosphorylation of Akt primarily due to a
reduction in total AKT protein levels. Metformin treatment also
inhibited mTOR phosphorylation (Fig.
5B).
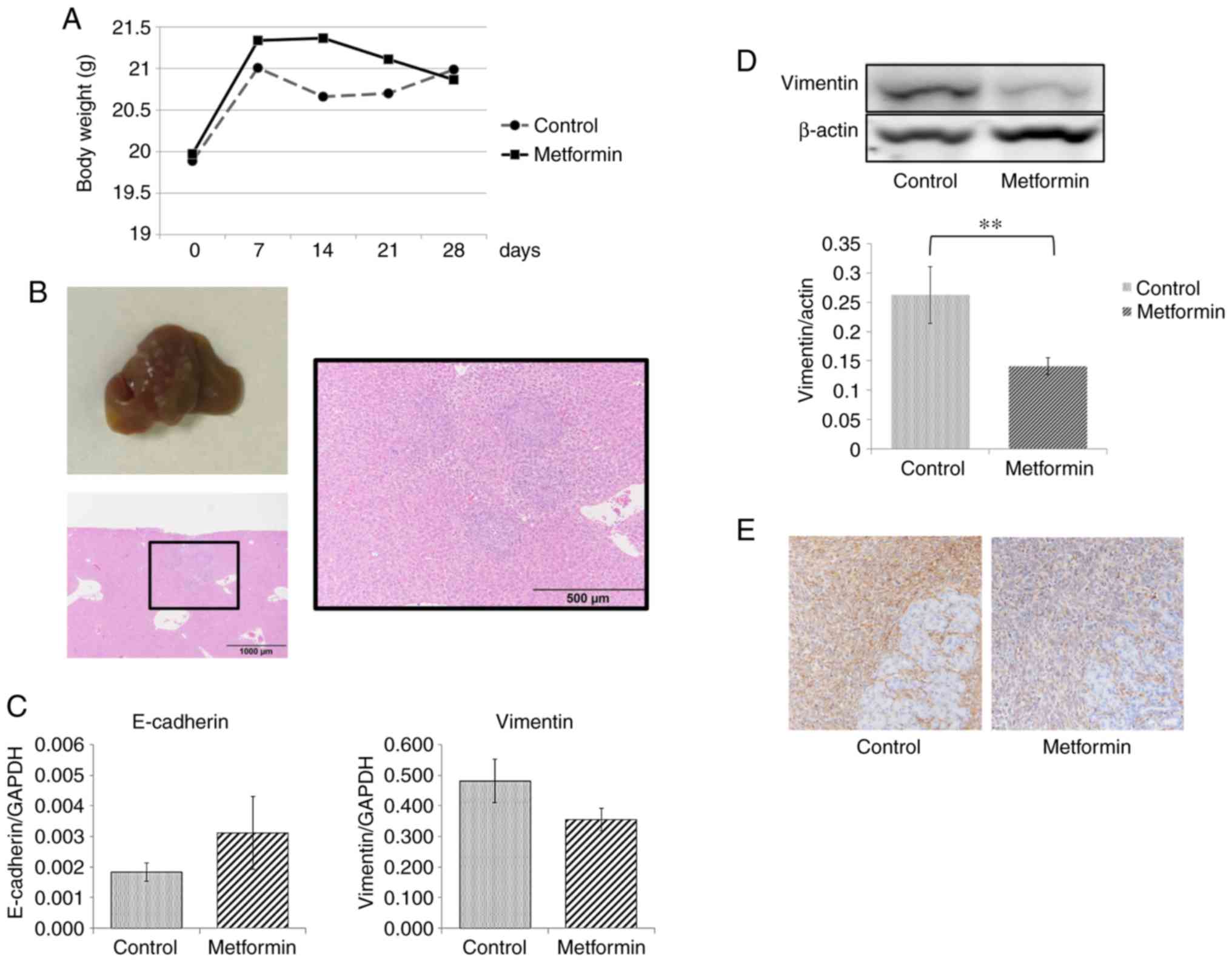
Metformin suppresses tumor growth and
metastasis in vivo
Pancreatic tumors survived and grew in the
pancreatic tails of all sacrificed mice in the present study. A
total of 2 mice in the metformin group and 1 mouse in the control
group were euthanized due to a >20% weight gain due to ascite
retention. A total of 2 mice in the metformin group died within 3
days following SOI, presumably due to suture failure. Consequently,
14 mice in the control group and 11 mice in the metformin group
were evaluated for metastasis. Primary pancreatic tumors tended to
be smaller in both size and weight in the metformin group than in
the control group, but this was not statistically significant
(Table I) (Fig. 6A). For evaluation of metastasis, we
observed liver and lung surfaces, and further sectioned to evaluate
the metastasis microscopically (Fig.
6B). Liver metastasis was observed in 15 out of 25 mice. All
mice with liver metastases had multiple tumors (range 3–20). In the
control group, liver metastasis was observed in 78.6% (11/14) of
the mice, whereas in the metformin group, metastasis was observed
in only 36.4% (4/11). This suppression of metastasis by metformin
treatment was statistically significant (P=0.049, Table I). Lung metastasis was observed in 5
out of 25 mice, and although the frequency of lung metastasis was
slightly higher in the control group (28.6% vs. 9.1%), it did not
reach statistical significance (Table
I). Subsequently, the levels of EMT-related markers in the
primary pancreatic tumors in 12 mice were measured. RT-qPCR
analysis revealed that E-cadherin expression tended to increase and
vimentin expression tended to decrease with metformin treatment
(Fig. 6C), although neither reached
statistical significance (P=0.07 and P=0.07, respectively). As
shown by western blot analysis and immunohistochemical analysis,
the expression of vimentin in the metformin group was significantly
lower than that in the control group (Fig. 6D and E).
 | Table I.Tumor growth and metastatic pattern
in mice administered metformin and control mice. |
Table I.
Tumor growth and metastatic pattern
in mice administered metformin and control mice.
|
| Control (n=14) | Metformin
(n=11) | P-value |
|---|
| Body weight at SOI
(g) | 19.9±1.2 | 20.0±0.9 | 0.835 |
| Body weight at
sacrifice (g) | 21.0±1.7 | 20.9±1.5 | 0.861 |
| Tumor growth |
|
|
|
|
yes | 14 | 11 | 1.000 |
| no | 0 | 0 |
|
| Tumor volume
(mm3) | 601.4±179.2 | 492.1±262.3 | 0.249 |
| Tumor weight
(mg) | 456.5±182.9 | 368.1±212.2 | 0.295 |
| Liver
metastasis |
|
|
|
|
yes | 11 | 4 | 0.049 |
| no | 3 | 7 |
|
| Lung
metastasis |
|
|
|
|
yes | 4 | 1 | 0.341 |
| no | 10 | 10 |
|
Discussion
EMT is a crucial biological process in cancer
invasion and metastasis. However, the details of the mechanisms of
EMT in pancreatic cancer and how this process can be suppressed
remain to be explored. The present study focused on whether
metformin, widely used in the treatment of type 2 diabetes
mellitus, can inhibit the EMT of pancreatic cancer cells. The
results indicated that metformin suppressed the TGF-β1-induced EMT
of pancreatic cells and inhibited liver metastasis in vivo.
Moreover, the results strongly suggested that metformin inhibited
EMT by inhibiting the TGF-β signaling pathways (i.e., the Smad2/3
and Akt/mTOR pathways). To the best of our knowledge, this is the
first study to demonstrate that metformin inhibits EMT and
metastasis formation by suppressing the intracellular TGF-β
signaling pathways in pancreatic cancer.
Recently, metformin has attracted attention due to
its anti-tumor effects (3). Strong
associations between metformin use and the reduced risk of several
cancer types, including pancreatic cancer have been found in
diabetic patients (4,23). Preclinical studies have suggested
that metformin can inhibit the growth of pancreatic cancer both
in vitro and in vivo (24,25).
Metformin systemically ameliorates hyperinsulinemia, hyperglycemia
and hyperlipidemia, which are involved in both cancer initiation
and progression. An antitumor effect of metformin is triggered when
several signals, such as phosphoinositide 3-kinase (PI3K) and MAPK
are suppressed (26–28). Metformin also inhibits the mTOR
pathway and its downstream substrates (29,30),
reduces cyclin D1 expression and cell cycle arrest in a
concentration-dependent manner (31), suppresses angiogenesis through the
reduction of vascular endothelial growth factor (VEGF) expression
(32), and induces apoptosis by p53
activation (3,33).
Recent studies have suggested that metformin
inhibits EMT in several types of cancer (13–19).
The results of these studies suggest that there are several
mechanisms through which metformin inhibits EMT. Zhao et al
reported that metformin inhibited interleukin (IL)-6-induced EMT in
colorectal cancer by blocking signal transducer and activator of
transcription 3 (STAT3) phosphorylation (17). Metformin has also been reported to
inhibit EMT in prostate cancer through the COX2/PGE2/STAT3 axis
(15). TGF-β1 acts as a potent
driver of cancer progression through the induction of EMT in
various types of cancer. In TGF-β-induced EMT, both the Smad and
non-Smad pathways (i.e., Akt/mTOR and MAP kinase pathways) are
activated, thereby increasing EMT-related gene expression (10,34–38).
Previous studies have demonstrated that metformin blocks the Smad
signaling pathway and inhibits TGF-β-induced EMT (19,39,40).
On the other hand, metformin has also been reported to block
TGF-β1-induced EMT through non-Smad pathways, such as the
Akt/mTOR/Zeb1 pathway (16). Thus,
there are several mechanisms responsible for the inhibition of EMT
by metformin, depending on the type of cell used, the EMT inducer,
and others.
To the best of our knowledge, until recently, there
were no studies available on the effect of metformin on EMT in
pancreatic cancer. However, Duan et al demonstrated that
metformin reduced TGF-β1 production in pancreatic cells, resulting
in the inhibition of autocrine TGF-β1 signaling, thereby inhibiting
EMT (41). However, the effect of
metformin on TGF-β1 signaling pathways was not investigated in this
previous study. In pancreatic cancer, there have been no studies
published to date on the effect of metformin on the TGF-β-induced
Smad and non-Smad signaling pathways involved in EMT, at least to
the best of our knowledge. The results of the present study
demonstrated that metformin inhibited TGF-β1-induced-EMT through
the downregulation of the Smad pathway in PANC-1 cells and the
downregulation of the Akt/mTOR pathway in BxPC-3 cells. These
findings suggest that metformin inhibits TGF-β1-induced-EMT through
the downregulation of different pathways, depending on the cell
line, and to the best of our knowledge, this is the first report to
demonstrate an inhibition of TGF-β1-induced-EMT by metformin
through the downregulation of intracellular TGF-β1 signaling
pathways in pancreatic cancer cells.
Of note, it was found that metformin inhibited EMT
in vitro and liver metastasis in vivo at clinical
doses. In the present study, metformin was administered to mice
daily by intraperitoneal injection at 125 mg/kg, which is
equivalent to the human dose of 600 mg/average size individual of
60 kg (42). Since the dose of
metformin for patients with type 2 diabetes mellitus is up to 2,250
mg/individual in Japan (43), it is
noteworthy that metformin has the potential to suppress cancer
metastasis at relatively low doses used clinically. In the present
study, an orthotopic tumor implantation model was used instead of a
spontaneous metastasis model. Focusing on the process of metastasis
formation, it was considered that this model would be more suitable
for the purposes of the present study to assess the effect of
metformin, which inhibits cancer cell proliferation, on cancer
metastasis. Furthermore, it was confirmed that metformin suppressed
liver metastasis without a significant decrease in the primary
pancreatic lesion.
There are a few limitations to the present study.
Although the assessment of motility is crucial for the assessment
of EMT, for the BxPC cells, this assay was not technically feasible
due to the potent adhesive force. Alternatively, other assays, such
as invasion assays, could overcome this limitation. For proteins
with a very low protein content, such as the phosphorylation of
Smad, mTOR and Akt, multiple western blot analyses could not be
performed as each assay required too many cells. As a result,
statistical analysis could not be performed. In in vivo
experiments, there were euthanized and dead mice in both groups
prior to the analysis for metastasis. Although the present study
demonstrated that metformin suppressed liver metastases, whether
the long-term use of metformin in tumor-bearing mice improves
survival needs to be confirmed in future studies. In addition, in
the present study, the number of metastases could not be accurately
compared due to the presence of a number of small metastatic foci.
The use of Pan02 cells engineered to express luciferase may
contribute to the accurate assessment of metastases.
In conclusion, the present study demonstrates that
metformin suppresses TGF-β1-induced EMT in pancreatic cells and
inhibits liver metastasis in vivo. Moreover, the results
demonstrated that the process of inhibiting EMT is mediated through
the Smad2/3 or Akt/mTOR pathway. Based on these findings, metformin
is expected to be clinically useful for the suppression of cancer
metastasis in patients with pancreatic cancer. To verify the
inhibitory effects of metformin on cancer metastasis, prospective
clinical studies, such as those in adjuvant settings are
necessary.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Grant-in-Aid
for Scientific Research (KAKENHI C) (17K09314) from the Japan
Society for the Promotion of Science (JSPS). YI received a research
fee from Takeda Pharmaceutical Co., Osaka, Japan, Sumitomo
Dainippon Pharmaceutical Co., Osaka Japan, Daiichi Sankyo Co.,
Tokyo Japan, and Pfizer, Tokyo, Japan. YN received lecture fees
from Takeda Pharmaceutical Co. Neither the funding agency nor any
outside organization participated in the study design or have any
competing interest.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article or are available from the
corresponding author on reasonable request.
Authors' contributions
TI, YN and YI conceived and designed the study and
modified the manuscript. JY collected and analyzed the data,
designed and performed the experiments, and drafted the manuscript.
JY, TI, TeO, NS, KI, KK, KU, and TT performed the cell culture and
wound healing assay. JY, TI, YE, SM, TaO, KM and KO performed
western blot analysis, RT-qPCR and in vivo experiments. JY
and YH performed the immunocytochemistry and immunohistochemistry
experiments. All authors participated in revising the manuscript,
read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the
Institutional Animal Care and Use Committee of Kyoto Prefectural
University of medicine under Assurance Number M30-618.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wolfgang CL, Herman JM, Laheru DA, Klein
AP, Erdek MA, Fishman EK and Hruban RH: Recent progress in
pancreatic cancer. CA Cancer J Clin. 63:318–348. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Andersen DK, Korc M, Petersen GM, Eibl G,
Li D, Rickels MR, Chari ST and Abbruzzese J: Diabetes,
pancreatogenic diabetes, and pancreatic cancer. Diabetes.
66:1103–1110. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
He H, Ke R, Lin H, Ying Y, Liu D and Luo
Z: Metformin, an old drug, brings a new era to cancer therapy.
Cancer J. 21:70–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Franciosi M, Lucisano G, Lapice E,
Strippoli GF, Pellegrini F and Nicolucci A: Metformin therapy and
risk of cancer in patients with type 2 diabetes: Systematic review.
PLoS One. 8:e715832013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang P, Li H, Tan X, Chen L and Wang S:
Association of metformin use with cancer incidence and mortality: A
meta-analysis. Cancer Epidemiol. 37:207–218. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bayraktar S, Hernadez-Aya LF, Lei X,
Meric-Bernstam F, Litton JK, Hsu L, Hortobagyi GN and
Gonzalez-Angulo AM: Effect of metformin on survival outcomes in
diabetic patients with triple receptor-negative breast cancer.
Cancer. 118:1202–1211. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wan G, Sun X, Li F, Wang X, Li C, Li H, Yu
X and Cao F: Survival benefit of metformin adjuvant treatment for
pancreatic cancer patients: A systematic review and meta-analysis.
Cell Physiol Biochem. 49:837–847. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Broadhurst PJ and Hart AR: Metformin as an
adjunctive therapy for pancreatic cancer: A review of the
literature on its potential therapeutic use. Dig Dis Sci.
63:2840–2852. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jie XX, Zhang XY and Xu CJ:
Epithelial-to-mesenchymal transition, circulating tumor cells and
cancer metastasis: Mechanisms and clinical applications.
Oncotarget. 8:81558–81571. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Okajima M, Kokura S, Ishikawa T, Mizushima
K, Tsuchiya R, Matsuyama T, Adachi S, Okayama T, Sakamoto N, Kamada
K, et al: Anoxia/reoxygenation induces epithelial-mesenchymal
transition in human colon cancer cell lines. Oncol Rep.
29:2311–2317. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Wu Z and Hu L: The regulatory
effects of metformin on the [SNAIL/miR-34]:[ZEB/miR-200] system in
the epithelial-mesenchymal transition(EMT) for colorectal
cancer(CRC). Eur J Pharmacol. 834:45–53. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Valaee S, Yaghoobi MM and Shamsara M:
Metformin inhibits gastric cancer cells metastatic traits through
suppression of epithelial-mesenchymal transition in a
glucose-independent manner. PLoS One. 12:e01744862017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tong D, Liu Q, Liu G, Xu J, Lan W, Jiang
Y, Xiao H, Zhang D and Jiang J: Metformin inhibits
castration-induced EMT in prostate cancer by repressing
COX2/PGE2/STAT3 axis. Cancer Lett. 389:23–32. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Song Y, Chen Y, Li Y, Lyu X, Cui J, Cheng
Y, Zhao L and Zhao G: Metformin inhibits TGF-beta1-induced
epithelial-to-mesenchymal transition-like process and stem-like
properties in GBM via AKT/mTOR/ZEB1 pathway. Oncotarget.
9:7023–7035. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao Z, Cheng X, Wang Y, Han R, Li L,
Xiang T, He L, Long H, Zhu B and He Y: Metformin inhibits the
IL-6-induced epithelial-mesenchymal transition and lung
adenocarcinoma growth and metastasis. PLoS One. 9:e958842014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakayama A, Ninomiya I, Harada S, Tsukada
T, Okamoto K, Nakanuma S, Sakai S, Makino I, Kinoshita J, Hayashi
H, et al: Metformin inhibits the radiation-induced invasive
phenotype of esophageal squamous cell carcinoma. Int J Oncol.
49:1890–1898. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li NS, Zou JR, Lin H, Ke R, He XL, Xiao L,
Huang D, Luo L, Lv N and Luo Z: LKB1/AMPK inhibits TGF-β1
production and the TGF-beta signaling pathway in breast cancer
cells. Tumour Biol. 37:8249–8258. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Uchiyama K, Naito Y, Takagi T, Mizushima
K, Hayashi N, Harusato A, Hirata I, Omatsu T, Handa O, Ishikawa T,
et al: Carbon monoxide enhance colonic epithelial restitution via
FGF15 derived from colonic myofibroblasts. Biochem Biophys Res
Commun. 391:1122–1126. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kimura-Tsuchiya R, Ishikawa T, Kokura S,
Mizushima K, Adachi S, Okajima M, Matsuyama T, Okayama T, Sakamoto
N, Katada K, et al: The inhibitory effect of heat treatment against
epithelial-mesenchymal transition (EMT) in human pancreatic
adenocarcinoma cell lines. J Clin Biochem Nutr. 55:56–61. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hwang HK, Murakami T, Kiyuna T, Kim SH,
Lee SH, Kang CM, Hoffman RM and Bouvet M: Splenectomy is associated
with an aggressive tumor growth pattern and altered host immunity
in an orthotopic syngeneic murine pancreatic cancer model.
Oncotarget. 8:88827–88834. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li D, Yeung SC, Hassan MM, Konopleva M and
Abbruzzese JL: Antidiabetic therapies affect risk of pancreatic
cancer. Gastroenterology. 137:482–488. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shi Y, He Z, Jia Z and Xu C: Inhibitory
effect of metformin combined with gemcitabine on pancreatic cancer
cells in vitro and in vivo. Mol Med Rep.
14:2921–2928. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kisfalvi K, Moro A, Sinnett-Smith J, Eibl
G and Rozengurt E: Metformin inhibits the growth of human
pancreatic cancer xenografts. Pancreas. 42:781–785. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Algire C, Amrein L, Bazile M, David S,
Zakikhani M and Pollak M: Diet and tumor LKB1 expression interact
to determine sensitivity to anti-neoplastic effects of metformin in
vivo. Oncogene. 30:1174–1182. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jalving M, Gietema JA, Lefrandt JD, de
Jong S, Reyners AK, Gans RO and de Vries EG: Metformin: Taking away
the candy for cancer? Eur J Cancer. 46:2369–2380. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schneider MB, Matsuzaki H, Haorah J,
Ulrich A, Standop J, Ding XZ, Adrian TE and Pour PM: Prevention of
pancreatic cancer induction in hamsters by metformin.
Gastroenterology. 120:1263–1270. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Muniraj T and Chari ST: Diabetes and
pancreatic cancer. Minerva Gastroenterol Dietol. 58:331–345.
2012.PubMed/NCBI
|
|
30
|
Yue W, Yang CS, DiPaola RS and Tan XL:
Repurposing of metformin and aspirin by targeting AMPK-mTOR and
inflammation for pancreatic cancer prevention and treatment. Cancer
Prev Res (Phila). 7:388–397. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ben Sahra I, Laurent K, Loubat A,
Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le
Marchand-Brustel Y and Bost F: The antidiabetic drug metformin
exerts an antitumoral effect in vitro and in vivo through a
decrease of cyclin D1 level. Oncogene. 27:3576–3586. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lund SS, Tarnow L, Stehouwer CD,
Schalkwijk CG, Teerlink T, Gram J, Winther K, Frandsen M, Smidt UM,
Pedersen O, et al: Impact of metformin versus repaglinide on
non-glycaemic cardiovascular risk markers related to inflammation
and endothelial dysfunction in non-obese patients with type 2
diabetes. Eur J Endocrinol. 158:631–641. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Buzzai M, Jones RG, Amaravadi RK, Lum JJ,
DeBerardinis RJ, Zhao F, Viollet B and Thompson CB: Systemic
treatment with the antidiabetic drug metformin selectively impairs
p53-deficient tumor cell growth. Cancer Res. 67:6745–6752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang YE: Non-Smad pathways in TGF-beta
signaling. Cell Res. 19:128–139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hoot KE, Lighthall J, Han G, Lu SL, Li A,
Ju W, Kulesz-Martin M, Bottinger E and Wang XJ:
Keratinocyte-specific Smad2 ablation results in increased
epithelial-mesenchymal transition during skin cancer formation and
progression. J Clin Invest. 118:2722–2732. 2008.PubMed/NCBI
|
|
36
|
Lamouille S and Derynck R: Cell size and
invasion in TGF-beta-induced epithelial to mesenchymal transition
is regulated by activation of the mTOR pathway. J Cell Biol.
178:437–451. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lamouille S, Connolly E, Smyth JW, Akhurst
RJ and Derynck R: TGF-β-induced activation of mTOR complex 2 drives
epithelial-mesenchymal transition and cell invasion. J Cell Sci.
125:1259–1273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Julien S, Puig I, Caretti E, Bonaventure
J, Nelles L, van Roy F, Dargemont C, de Herreros AG, Bellacosa A
and Larue L: Activation of NF-kappaB by Akt upregulates Snail
expression and induces epithelium mesenchyme transition. Oncogene.
26:7445–7456. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shin HS, Ko J, Kim DA, Ryu ES, Ryu HM,
Park SH, Kim YL, Oh ES and Kang DH: Metformin ameliorates the
phenotype transition of peritoneal mesothelial cells and peritoneal
fibrosis via a modulation of oxidative stress. Sci Rep. 7:56902017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Thakur S, Viswanadhapalli S, Kopp JB, Shi
Q, Barnes JL, Block K, Gorin Y and Abboud HE: Activation of
AMP-activated protein kinase prevents TGF-β1-induced
epithelial-mesenchymal transition and myofibroblast activation. Am
J Pathol. 185:2168–2180. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Duan W, Qian W, Zhou C, Cao J, Qin T, Xiao
Y, Cheng L, Li J, Chen K, Li X, et al: Metformin suppresses the
invasive ability of pancreatic cancer cells by blocking autocrine
TGF-β1 signaling. Oncol Rep. 40:1495–1502. 2018.PubMed/NCBI
|
|
42
|
Tan XL, Bhattacharyya KK, Dutta SK, Bamlet
WR, Rabe KG, Wang E, Smyrk TC, Oberg AL, Petersen GM and
Mukhopadhyay D: Metformin suppresses pancreatic tumor growth with
inhibition of NFkappaB/STAT3 inflammatory signaling. Pancreas.
44:636–647. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Odawara M, Kawamori R, Tajima N, Iwamoto
Y, Kageyama S, Yodo Y, Ueki F and Hotta N: Long-term treatment
study of global standard dose metformin in Japanese patients with
type 2 diabetes mellitus. Diabetol Int. 8:286–295. 2017. View Article : Google Scholar : PubMed/NCBI
|