Introduction
CD9, a member of the tetraspanin superfamily, is a
cell surface glycoprotein with a molecular weight of 24–27 kDa
(1). Recent studies have shown that
CD9 is expressed in 72% of adult B-lineage acute lymphoblastic
leukemia (B-ALL) (2) and 78% of
pediatric B-ALL (3) and correlates
with leukemia progression and clinical survival. Initially in 2009
and 2011, Nishida et al and Yamazaki et al reported
that the CD9 antigen-positive (CD9+) B-ALL cell
population possessed stem cell characteristics within the clone;
CD9+ B-ALL cells were relatively more resistant to
chemotherapeutic agents than CD9− cells (4,5). In 2015,
Arnaud et al identified that CD9 promoted the activation of
Ras-related C3 botulinum toxin substrate 1 (RAC1) and enhanced
C-X-C motif chemokine receptor 4-mediated migration and engraftment
of B-ALL cells to the bone marrow or testis (6). In 2018, Liang et al demonstrated
that CD9+ acute lymphoblastic leukemia (ALL) patients
exhibited a higher positive rate of the BCR-ABL fusion gene
compared with CD9− patients, and CD9 expression was
related to poor prognosis in ALL patients (2). In 2019, Leung et al reported that
CD9+ B-ALL children had a significantly lower 5-year
relapse-free survival rate than CD9− patients; the
administration of anit-CD9 antibody suppressed leukemia progression
in NOD/SCID mice xenografted with CD9+ cell lines and
primary B-ALL blasts from high-risk and refractory patients; the
blockage of CD9 inhibited B-ALL cell proliferation, induced cell
cycle arrest and promoted chemotherapeutic agent-induced apoptosis
(3). Furthermore, our recent study
demonstrated that the downregulation of CD9 inhibited cell
proliferation, adhesion, migration and invasion, while increasing
apoptosis and the cytotoxicity of chemotherapeutic agents in B-ALL
SUP-B15 cells (7). Therefore, CD9
serves an important role in the disease progression and clinical
prognosis in B-ALL. Nevertheless, the corresponding mechanisms
remain to be explored.
It is well known that the activation of the
phosphatidylinositol 3-kinase (PI3K)/serine/threonine-specific
protein kinase (AKT) signaling pathway plays an important role in
tumorigenesis and development. The PI3K/AKT signaling pathway has
been shown to be involved in a variety of tumor-related biological
functions, such as cell survival, apoptosis resistance, drug
resistance and metastasis (8).
Emerging evidence has also revealed that tetraspanin proteins could
regulate the PI3K/AKT signaling pathway. For example, the integrin
α3β1-tetraspanin protein complexes have been implicated in actin
cytoskeletal reorganization via a PI3K-dependent mechanism
(9). In addition, the ectopic
expression of the tetraspanin CD151 had a negative regulatory
effect on AKT activation (10).
Furthermore, it has been reported that interleukin (IL)-16 could
activate the PI3K signaling pathway by binding to CD9 in mast cells
(11). Considering the above
findings, it is hypothesized that CD9 might be involved in the
regulation of the biological behavior of B-ALL cells through the
PI3K/AKT pathway.
Therefore, the aims of the present study were: i) To
investigate the relationship between CD9 and the PI3K/AKT pathway;
and ii) to assess the in vitro anti-leukemia effects
mediated by inhibition of the PI3K/AKT pathway in CD9+
B-ALL cells.
Materials and methods
Cell lines and culture conditions
Cell lines SUP-B15 and 293T were purchased from The
American Type Culture Collection (ATCC). The SUP-B15 cell line was
authenticated by short tandem repeat DNA profiling analysis at
Suzhou Genetic Testing Biotechnology Corporation, China. SUP-B15
cells were cultured in Iscove's modified Dulbecco's medium (IMDM)
with 20% fetal bovine serum (FBS), and 293T cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) with 10% FBS. All cells
were maintained in a humidified incubator at 37°C in an atmosphere
of 5% CO2.
Short hairpin RNA (shRNA) lentiviral
transduction
The transduction protocols were described in our
previous paper (7). Briefly, 12 µg
PHY-310 lentiviral vector (hU6-MCS-CMV-ZsGreen1-PGK-Puro; Shanghai
Hanyin Biotechnology Co., Ltd.) containing an shRNA targeting
CD9 [the target sequence for CD9 shRNA:
5′-AGGAAGTCCAGGAGTTTTA-3′ (synthesized by Shanghai Hanyin
Biotechnology Co., Ltd.); primers used for targeting the
interference sequence of the CD9 gene: F:
GATCCAGGAAGTCCAGGAGTTTTATTCAAGAGATAAAACTCCTGGACTTCCTTTTTTTG and R:
AATTCAAAAAAAGGAAGTCCAGGAGTTTTATCTCTTGAATAAAACTCCTGGACTTCCTG) and 9
µg lentiviral packaging vector LV-PV001 [a second-generation
packaging vector; Han Yin Biotechnology (Shanghai) Co., Ltd.] were
mixed with polyethylenimine (Sigma-Aldrich; Merck KGaA) and
incubated for 20 min at 37°C. Then, the subconfluent 293T cells
(1.5×108/dish) in a 10-cm culture dish were transfected
with the transfection mixture. The blank PHY-310 vector (in order
to exclude the effect of the lentiviral vector on the experimental
results) was used as a negative control. At 48 h after
transfection, lentiviral particles produced from the transfected
293T cells were harvested and purified by ultra-centrifugation at
3,000 × g for 2.5 h at 4°C. SUP-B15 cells were transduced with the
lentiviral particles [multiplicity of infection (MOI)=100)] by
centrifugation in the presence of 8 µg/ml polybrene (Sigma-Aldrich;
Merck KGaA) at 37°C for 4 h, and then stable cell lines were
generated by selection with puromycin (1 µg/ml) for 48 h. The
efficiency of CD9 knockdown was evaluated by reverse
transcription-quantitative PCR (RT-qPCR). The method of RT-qPCR was
carried out according to a previous study (7).
Enzyme linked immunosorbent assay
(ELISA)
The levels of phosphorylated-PI3K (p-PI3K) in
SUP-B15 cells were measured using the Human p-PI3K ELISA kit (cat.
no. M1060625; Shanghai Enzyme-linked Biotechnology Co., Ltd.)
according to the manufacturer's protocol. Briefly, cells were lysed
by three consecutive freeze-thawing cycles (10 min each), and then
centrifuged at 860 × g for 10 min at 4°C. The total protein
concentration in the supernatant was determined using a BCA protein
assay kit (Beyotime Institute of Biotechnology). Subsequently, 50
µl of total protein extraction (1:5 dilution with PBS) or standards
along with 50 µl of biotin-labeled anti-p-PI3K antibody were
incubated on an antibody-coated plate for 1 h at 37°C. Wells were
then washed three times. The next step involved adding 80 µl of
streptavidin-labeled horseradish peroxidase (HRP) to each well,
followed by incubation at 37°C for 30 min. After washing for three
times, 50 µl of substrate solution A along with 50 µl of substrate
solution B was added to each well and the plate was incubated at
37°C for 10 min. The reaction was quenched by the addition of 50 µl
of stop solution. The plate was analyzed by evaluating the
absorbance at 450 nm using a Spectra MAX M5 microplate reader
(Molecular Devices).
Western blot analysis
The methods of protein extraction and western
blotting were carried out as previously described (7). Briefly, cells were lysed using RIPA
lysis buffer (Beyotime Institute of Biotechnology) and the total
protein concentration was measured by the BCA protein assay kit
(Beyotime Institute of Biotechnology). Total protein was then
separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred to a PVDF membrane. The
membranes were blocked with 5% skim milk for 1 h at room
temperature, and sequentially incubated overnight with primary
antibodies at 4°C. The primary antibodies used were
anti-phosphorylated-AKT (anti-p-AKT; 1:1,000 dilution, cat. no.
9018; Cell Signaling Technology, Inc.), anti-AKT (1:1,000 dilution,
cat. no. 2938; Cell Signaling Technology, Inc.), anti-p53 (1:1,000
dilution, cat. no. CY5131; Abways, Inc.), anti-p21 (1:1,000
dilution, cat. no. CY5088; Abways, Inc.), anti-cleaved caspase 3
(1:1,000 dilution, cat. no. CY5051; Abways, Inc.),
anti-P-glycoprotein (anti-P-gp; 1:1,000 dilution, cat. no. 13978;
Cell Signaling Technology, Inc.), anti-multidrug-resistance
associated protein 1 (anti-MRP1; 1:1,000 dilution, cat. no. CY6878;
Abways, Inc.), anti-breast cancer resistance protein (anti-BCRP;
1:1,000 dilution, cat. no. 10051-1-AP, ProteinTech Group, Inc.),
anti-matrix metalloproteinase 2 (anti-MMP2; 1:1,000 dilution, cat.
no. BS1236; Bioworld Technology, Inc.), anti-phosphorylated-focal
adhesion kinase (anti-p-FAK; 1:1,000 dilution, cat. no. CY6207;
Abways, Inc.), anti-FAK (1:1,000 dilution, cat. no. CY5464; Abways,
Inc.) and anti-β-actin (1:5,000 dilution, cat. no. ab2001; Abways,
Inc.). Following incubation using HRP-conjugated anti-rabbit
antibody (1:10,000 dilution, cat. no. 7074P2; Cell Signaling
Technology, Inc.) or HRP-conjugated anti-mouse antibody (1:10,000
dilution, cat. no. 7076; Cell Signaling Technology, Inc.) at room
temperature for 2 h, the protein bands were analyzed with
chemiluminescent solution (ECL Western Blotting Detection Reagents;
GE Healthcare Life Sciences). The relative level of protein
expression was quantified by ImageJ software (version 1.49p;
National Institutes of Health) and standardized to β-actin
levels.
Cell proliferation assay
The cell proliferation was determined by use of a
Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.).
Cells were cultured in 96-well microplates at a density of
3×103 cells/well. After incubation at 37°C for 24, 48,
72, 96 and 120 h, 10 µl of CCK-8 solution was added to each well
and the cells were incubated at 37°C for 2 h. The absorbance was
measured at 450 nm using a Spectra MAX M5 microplate reader
(Molecular Devices).
Drug sensitivity assay
The measurement of in vitro drug sensitivity
to chemotherapeutic agents were carried out according to our
previous study (7). Cells were
exposed to various concentrations of chemotherapeutic agents,
including vincristine (VCR; Selleck Chemicals), daunorubicin (DNR;
MedChemExpress), cyclophosphamide (CPM; MedChemExpress) and
dexamethasone (DXM; Selleck Chemicals), at 37°C for 48 h, and then
CCK-8 assay was used to detect the cell viability. The optical
density (OD) was measured at 450 nm using a Spectra MAX M5
microplate reader (Molecular Devices). The percentage of
cytotoxicity was calculated by the following formula: Cytotoxicity
(%)=(1-mean OD of treated/mean OD of control) ×100.
Cell adhesion assay
An artificial basement membrane was prepared by
adding 0.5 µg Superfibronectin (Sigma-Aldrich; Merck KGaA) into
each well of a 96-well plate and incubating at 4°C overnight.
Subsequently, a total of 1×105 cells resuspended in 200
µl IMDM medium was seeded into each well of a 96-well plate and
allowed to adhere to the Superfibronectin at 37°C for 90 min in a
humidified 5% CO2 atmosphere. Non-adherent cells were
removed by rinsing with PBS and adherent cells were then quantified
by CCK-8 assay.
Cell migration and invasion
assays
For the cell migration assay, a total of
1×104 cells in serum-free IMDM medium (100 µl) was
placed in the upper chamber of an 8-µm pore size Transwell plate
(Corning, Inc.) and 800 µl of IMDM medium containing 10% FBS was
added to the lower chamber as a chemoattractant. After incubation
for 72 h at 37°C in a 5% CO2 atmosphere, cells that
migrated to the lower chamber were harvested and stained with 1%
crystal violet at room temperature for 30 min after fixation with
4% paraformaldehyde at room temperature for 20 min. Finally, a
hemocytometer was used to count the cells under a light microscope
(magnification, ×100). The invasion assay was performed using the
same method as the migration assay, except for the precoating of
the upper chamber with 50 µl Matrigel (1 mg/ml; BD Biosciences) at
37°C for 5 h.
Plasmid construction
The full-length cDNA of CD9 was cloned into
the mammalian expression plasmid pcDNA3.1-Flag-MYC (Gene
Universal), forming a MYC tagged CD9 expression vector
pcDNA3.1-MYC-CD9, while full-length cDNA of PI3K-p85α or PI3K-p85β
was inserted into pcDNA3.1-Flag-HA (Gene Universal), forming a HA
tagged PI3K-p85α expression vector pcDNA3.1-HA-p85α or PI3K-p85β
expression vector pcDNA3.1-HA-p85β. The empty plasmids,
pcDNA3.1-Flag-MYC and pcDNA3.1-Flag-HA, were used as negative
control groups. Vector pET28 (Gene Universal) was used to construct
vectors expressing glutathione S-transferase (GST)-p85α or GST-p85β
fusion proteins.
GST pull-down assay
MYC-CD9 expression vector (2.5 µg) was mixed with
Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific,
Inc.) and incubated for 20 min at 37°C. Then, 293T cells in 10-cm
dishes were grown to 90% confluence and transfected with the
mixture. At 48 h after transfection, the cells were harvested and
lysed by RIPA lysis buffer (Beyotime Institute of Biotechnology).
After centrifugation at 16,560 × g for 15 min at 4°C, the
supernatants were isolated. On the other hand, BL-21 bacterial
cells were transformed with plasmids encoding the GST-tagged
proteins and then grown at 37°C until reaching log phase. GST
protein expression was induced by incubation with
isopropyl-1-thio-β-galactopyranoside (IPTG; 1 mmol/l) for 6 h. In
order to purify the GST fusion proteins, cells were lysed by
sonication in lysis buffer [50 mmol/l Tris (pH 7.4), 150 mmol/l
NaCl and 1% NP-40], and the resulting lysate was incubated for 1 h
at 4°C with glutathione-agarose beads (cat. no. G4510;
Sigma-Aldrich; Merck KGaA). The beads were harvested by
centrifugation and washed with lysis buffer.
For CD9 binding, 50 µl of whole cell lysate from
transfected 293T cells were incubated for 48 h with the GST-tagged
proteins bound to beads. Unbound CD9 protein was removed by three
washes with lysis buffer (1 ml each). Bound proteins were eluted by
boiling in 1X loading buffer, separated by 10% SDS-PAGE, and
examined by immunoblot analysis with anti-MYC antibody (1:1,000
dilution; cat. no. A7470; Sigma-Aldrich; Merck KGaA).
Co-immunoprecipitation assay
MYC-CD9 expression vector (2.5 µg) and either 2.5 µg
of HA-PI3K-p85α or HA-PI3K-p85β expression vector were mixed with
Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific,
Inc.) and incubated for 20 min at 37°C. Then, 293T cells in 10-cm
dishes were grown to 70–90% confluence and transfected with the
mixture. At 48 h after transfection, the cells were harvested and
lysed by RIPA lysis buffer (Beyotime Institute of Biotechnology).
Following the centrifugation using the same method described
previously, the supernatants were isolated and immunoprecipitated
with 5 µl anti-MYC antibody (cat. no. A7470; Sigma-Aldrich; Merck
KGaA). Detection of the co-precipitated HA-PI3K-p85α or
HA-PI3K-p85β was performed using western blotting by incubating
with an anti-HA antibody (1:1,000 dilution, cat. no. ab49969;
Abcam, Inc.). The supernatants described above were also subjected
to direct western blot analysis to confirm the expression of
MYC-CD9 and either HA-PI3K-p85α or HA-PI3K-p85β.
In vitro anti-leukemia effects of the
PI3K/AKT pathway inhibitor in SUP-B15 cells
SUP-B15 cells (5×103) were treated with
the PI3K/AKT pathway inhibitor LY294002 (0.6 µmol/l; cat. no.
HY-10108, MedChemExpress) at 37°C for 24 h, and then subjected to
cell proliferation, drug sensitivity, adhesion, migration,
invasion, ELISA and western blot assays, respectively.
Statistical analysis
Data are expressed as the mean ± standard deviation
(SD) from at least three independent experiments. Statistical
significance among groups was assessed with unpaired Student's
t-test and two-way ANOVA followed by Sidak's multiple comparisons
test. A P-value of <0.05 was considered as indicative of a
statistically significant difference. All statistical analyses were
performed using GraphPad Prism version 8.0.1 (GraphPad Software,
Inc.).
Results
CD9 activates PI3K/AKT signaling
To study the effect of CD9 on the PI3K/AKT pathway,
we assessed activation of the pathway in a CD9-knockdown B-ALL cell
line SUP-B15. We used the lentiviral-mediated shRNA approach in
SUP-B15 cells to establish an efficient permanent method of
downregulating the expression of CD9. Lentiviral delivery of shRNA
targeted against CD9 led to a significantly downregulation
of CD9 mRNA in SUP-B15 cells, as measured by RT-qPCR
(Fig. S1). ELISA results showed that
the level of p-PI3K protein was significantly reduced in the
SUP-B15 cells transduced with the PHY-310 lentiviral vector
containing shRNA targeting CD9 (SUP-B15-shCD9 group)
compared with wide-type SUP-B15 cells (SUP-B15-WT group) and those
cells transduced with a blank PHY-310 vector (SUP-B15-shControl
group; Fig. 1A). In addition, western
blot analysis revealed a significant reduction in the p-AKT/AKT
ratio in SUP-B15 cells after CD9 knockdown (Fig. 1B and C). In addition to p-PI3K and
p-AKT, we also tested their downstream targets, including drug
resistance-(such as P-gp, MRP1 and BCRP) and cell motility-related
molecules (such as MMP2 and p-FAK), by western blotting in SUP-B15
cells after CD9 knockdown. Silencing of CD9
significantly decreased P-gp, MRP1, BCRP, MMP2 expression and the
p-FAK/FAK ratio (Fig. 1B and C).
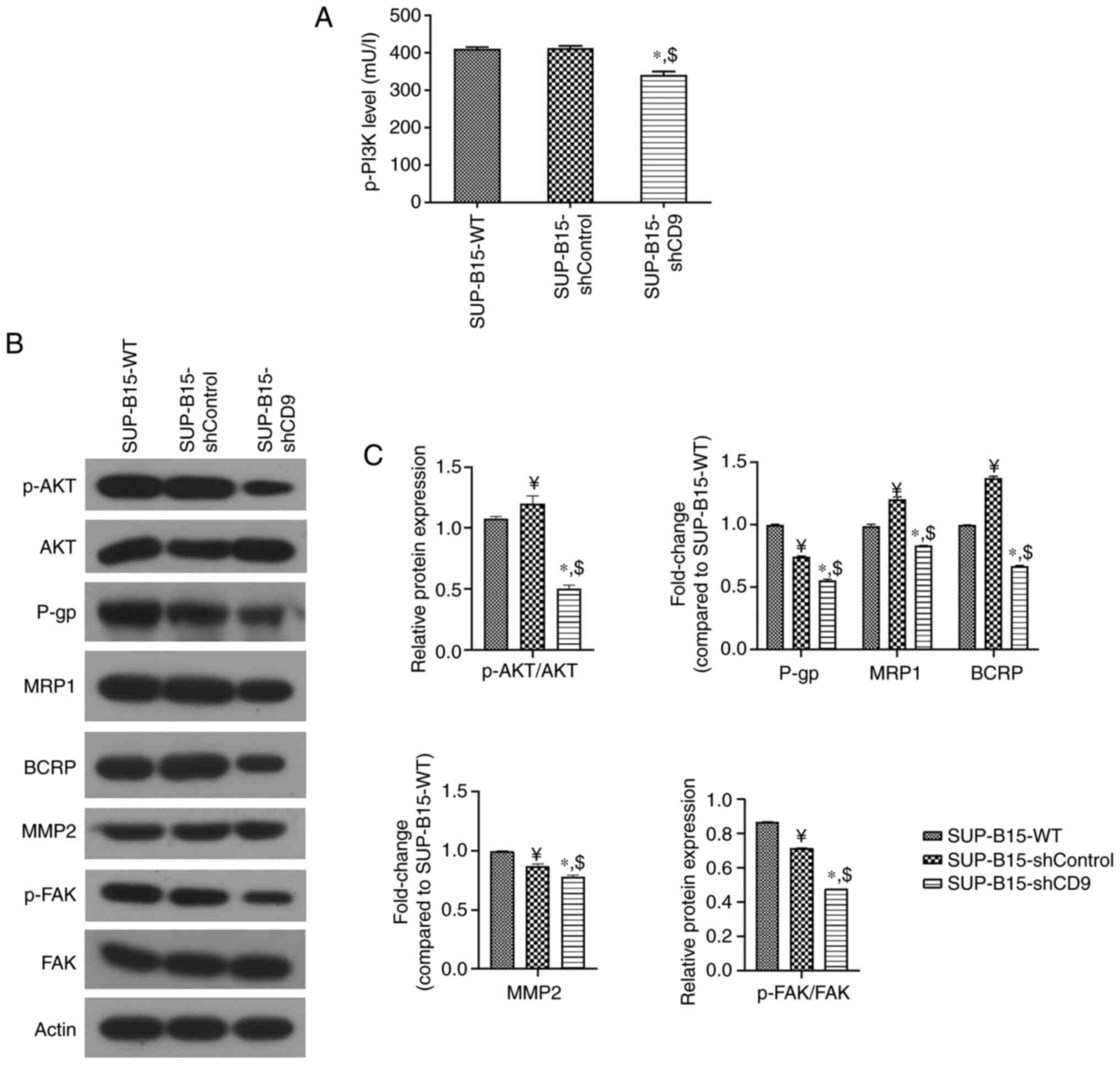 | Figure 1.CD9 knockdown suppresses the
PI3K/AKT pathway. (A) ELISA was used to measure the expression of
p-PI3K. (B) Western blot analysis was used to detect the protein
levels of p-AKT, as well as drug-resistance-related (P-gp, MRP1 and
BCRP) and motility-related factors (MMP2 and p-FAK). β-actin was
used as the loading control. (C) Densitometry results are expressed
as fold change against untreated control (normalized to the density
of the corresponding β-actin band), or the ratio of phosphoprotein
to total protein. SUP-B15-WT represents the wild-type SUP-B15
cells; SUP-B15-shControl represents the SUP-B15 cells transduced
with a blank PHY-310 vector; SUP-B15-shCD9 represents the SUP-B15
cells transduced with the PHY-310 lentiviral vector containing
shRNA targeting CD9; ¥P<0.05 vs. SUP-B15-WT
group; *P<0.05 vs. SUP-B15-WT group; $P<0.05 vs.
SUP-B15-shControl group. p-PI3K,
phosphorylated-phosphatidylinositol-3 kinase; p-AKT,
phosphorylated-protein kinase B; P-gp, P-glycoprotein; MRP1,
multidrug resistance-associated protein 1; BCRP, breast cancer
resistance protein; MMP2, matrix metalloproteinase 2; p-FAK,
phosphorylated-focal adhesion kinase. |
CD9 interacts directly with
PI3K-p85
To confirm the interaction between CD9 and PI3K-p85,
we constructed GST-PI3K-p85α and GST-PI3K-p85β prokaryotic
expression vectors, as well as MYC-CD9 mammalian expression vector.
Using these vectors we assessed the interaction between CD9 protein
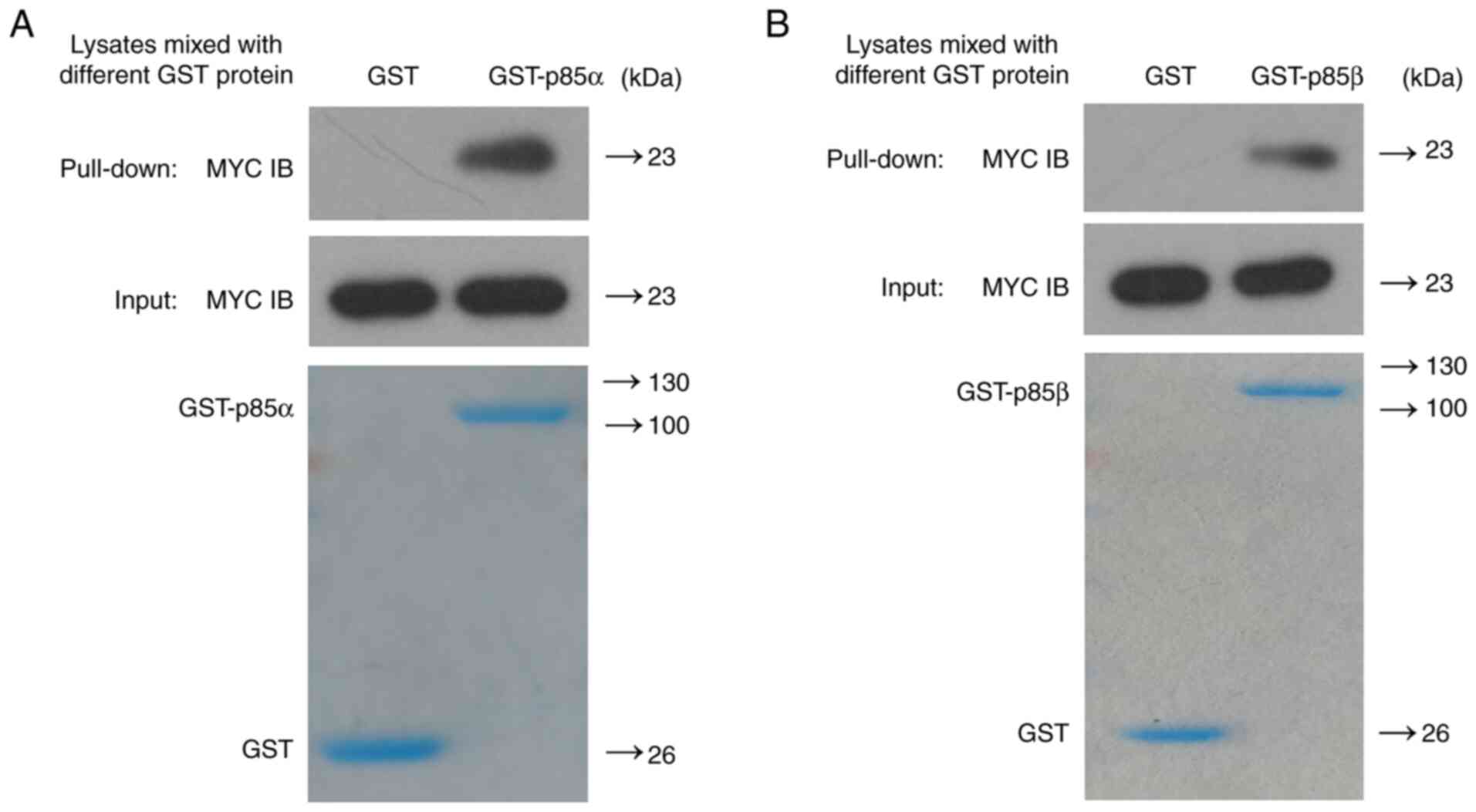
and either PI3K-p85α or PI3K-p85β by GST pull-down assay.
Successful transfection of 293T cells with the CD9
expression plasmid was confirmed by western blot analysis (Fig. S2). As shown in Fig. 2, MYC-CD9 recombinant protein was able
to bind to either GST-PI3K-p85α or GST-PI3K-p85β fusion protein
instead of the GST control protein, suggesting that CD9 directly
binds to both PI3K-p85α and PI3K-p85β in vitro.
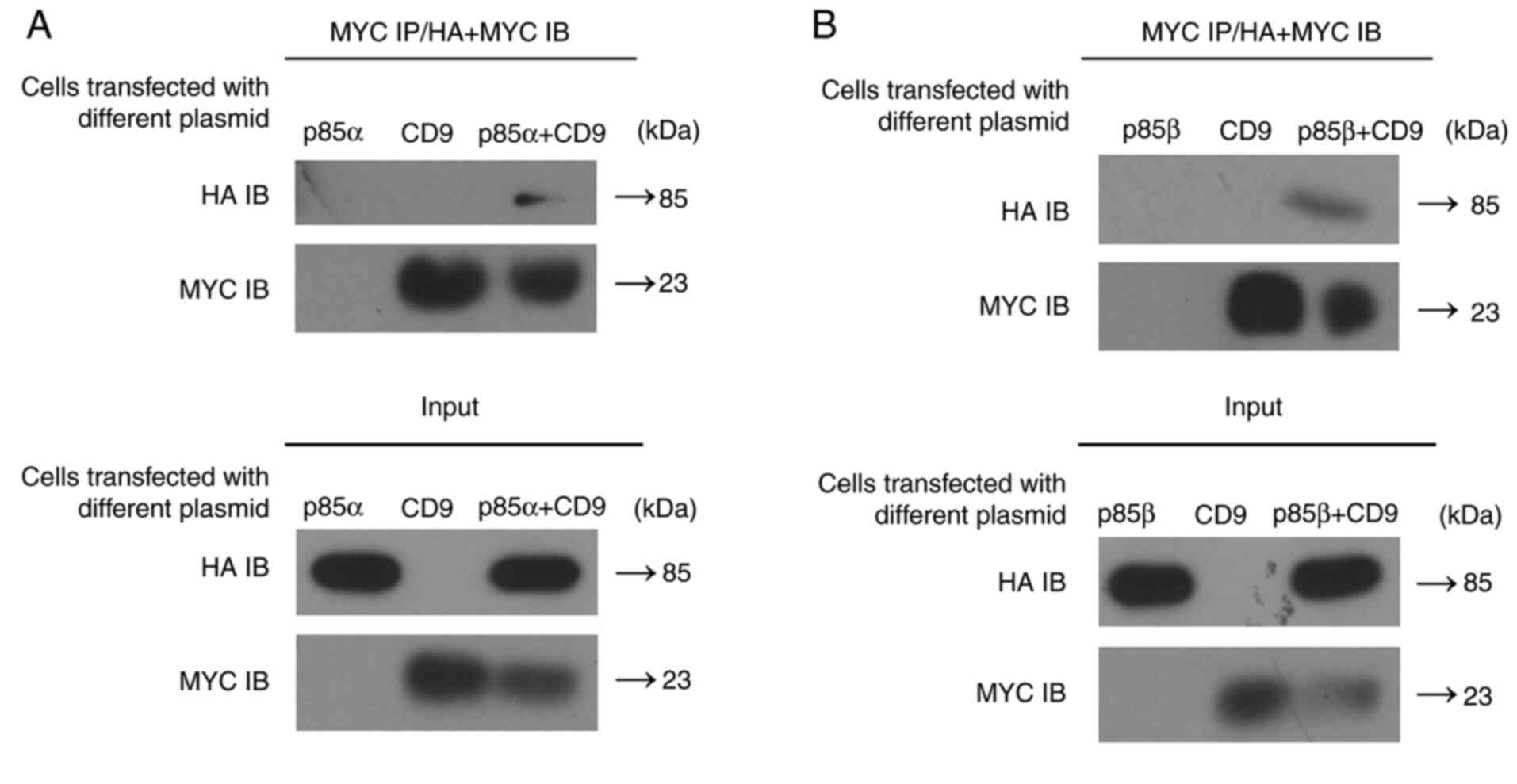
Since CD9 interacts directly with both PI3K-p85α and
PI3K-p85β in vitro, we next investigated whether CD9 binds
to PI3K-p85 in vivo. We constructed expression vectors for
MYC-tagged CD9 as well as HA-tagged PI3K-p85α or PI3K-p85β.
Successful transfection of 293T cells with PI3K-p85α
expression plasmid or PI3K-p85β expression plasmid was
confirmed by western blot analysis (Figs. S3 and 4). Then, MYC-CD9 expression vector and
either the HA-PI3K-p85α or HA-PI3K-p85β expression vector were
transfected into 293T cells and the cell extracts were subjected to
the co-immunoprecipitation assay using an anti-MYC antibody. Both
PI3K-p85α and PI3K-p85β were detected in immune complexes of
MYC-CD9 (Fig. 3), supporting that CD9
interacts with both PI3K-p85α and PI3K-p85β in vivo.
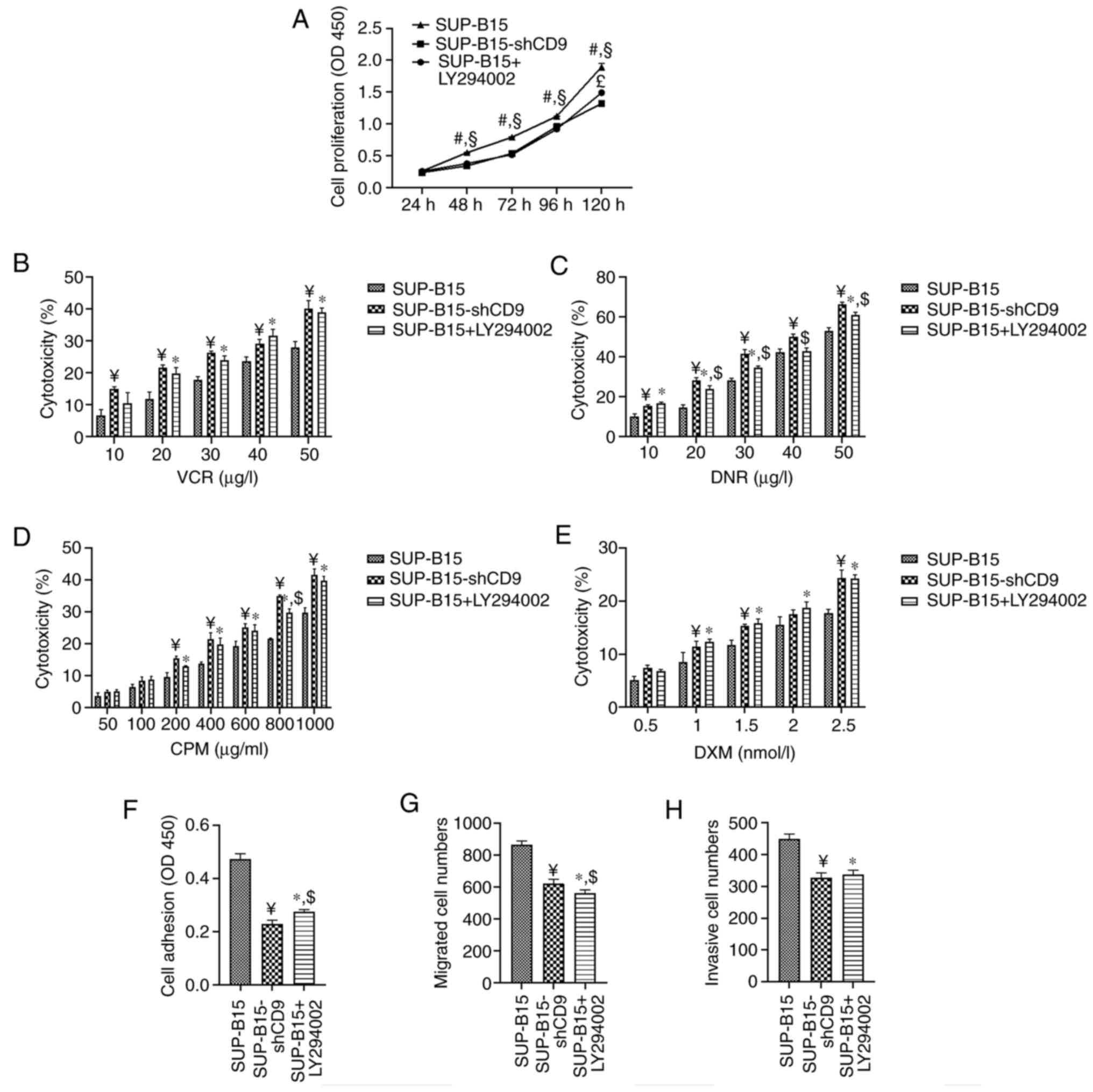 | Figure 4.Treatment with PI3K/AKT inhibitor
LY294002 suppresses cell proliferation, adhesion, migration and
invasion, while increasing the cytotoxicity of chemotherapeutic
agents in SUP-B15 cells. (A) Cell proliferation curves of SUP-B15
cells were measured by a CCK-8 assay. (B-E) SUP-B15 cells were
treated with different concentration gradient of (B) VCR, (C) DNR,
(D) CPM and (E) DXM for 48 h. CCK-8 assay was used to measure cell
viability. (F) The adhesion ability of SUP-B15 cells was evaluated
by cell adhesion to Superfibronectin. Adherent cells were
quantified by CCK-8 assay. (G) The migration and (H) invasion
ability of SUP-B15 cells was measured by the Transwell assay with
uncoated or Matrigel-coated membranes. Migrated and invasive cells
were counted by hemocytometer under a light microscope.
SUP-B15-shCD9 represents the SUP-B15 cells transduced with the
PHY-310 lentiviral vector containing shRNA targeting CD9;
#P<0.05 vs. SUP-B15-shCD9 cells without LY294002
treatment; §P<0.05 vs. SUP-B15 cells treated with
LY294002; £P<0.05 vs. SUP-B15-shCD9 cells without
LY294002 treatment; ¥P<0.05 vs. SUP-B15 cells without
LY294002 treatment; *P<0.05 vs. SUP-B15 cells without LY294002
treatment; $P<0.05 vs. SUP-B15-shCD9 cells without
LY294002 treatment; CCK-8, Cell Counting Kit-8; VCR, vincristine;
DNR, daunorubicin; CPM, cyclophosphamide; DXM, dexamethasone; OD,
optical density. |
The PI3K/AKT pathway inhibitor has in
vitro anti-leukemia effects in SUP-B15 cells
Since CD9 is involved in the regulation of the
biological behavior of B-ALL cells through the PI3K/AKT pathway, we
furthermore investigated the anti-leukemia effects of the
inhibition of the PI3K/AKT pathway in SUP-B15 cells using LY294002,
a PI3K/AKT pathway inhibitor. The results showed that the PI3K/AKT
inhibitor LY294002 mirrored the effects of CD9 knockdown in
SUP-B15 cells. Treatment with LY294002 inhibited cell proliferation
(Fig. 4A) as well as increased the
inhibitory response of SUP-B15 cells to VCR (Fig. 4B), DNR (Fig.
4C), CPM (Fig. 4D) and DXM
(Fig. 4E), respectively. In addition,
treatment with LY294002 inhibited adhesion (Fig. 4F), migration (Fig. 4G) and invasion (Fig. 4H) of SUP-B15 cells. To test the effect
of LY294002 on the expression of p-PI3K, ELISA was performed after
exposure to the compound for 24 h. The protein level of p-PI3K was
significantly reduced after treatment with LY294002 (Fig. 5A). Western blot assay also showed a
reduction in the p-AKT/AKT ratio by LY294002 treatment (Fig. 5B and C), suggesting that PI3K/AKT
signaling was inhibited. Additionally, the treatment with LY294002
increased the protein levels of p53, p21 and cleaved caspase-3,
while decreasing P-gp, MRP1, BCRP, MMP-2 expression and the
p-FAK/FAK ratio (Fig. 5B and C).
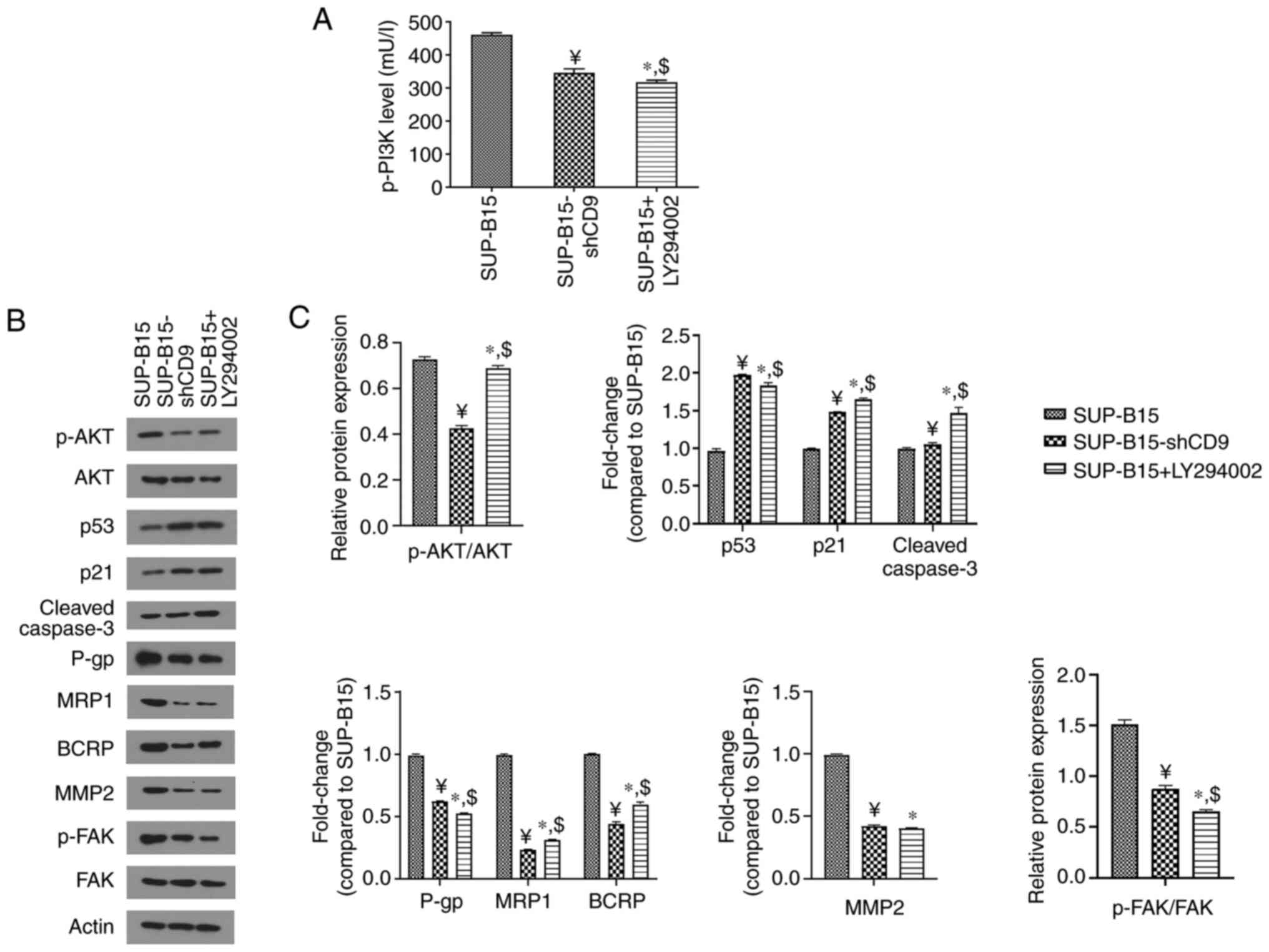 | Figure 5.The treatment with PI3K/AKT inhibitor
LY294002 suppresses the PI3K/AKT pathway. (A) ELISA was used to
measure the expression of p-PI3K. (B) Western blot analysis was
used to detect the protein levels of p-AKT, as well as cell cycle-
and apoptosis-related (including p53, p21 and cleaved caspase-3),
drug-resistance-related (including P-gp, MRP1 and BCRP) and
motility-related factors (including MMP2 and p-FAK). β-actin was
used as the loading control. (C) Densitometry results are expressed
as fold change against untreated control (normalized to the density
of the corresponding β-actin band), or the ratio of phosphoprotein
to total protein. SUP-B15-shCD9 represents the SUP-B15 cells
transduced with the PHY-310 lentiviral vector containing shRNA
targeting CD9; ¥P<0.05 vs. SUP-B15 cells
without LY294002 treatment; *P<0.05 vs. SUP-B15 cells without
LY294002 treatment; $P<0.05 vs. SUP-B15-shCD9 cells
without LY294002 treatment. p-PI3K,
phosphorylated-phosphatidylinositol-3 kinase; p-AKT,
phosphorylated-protein kinase B; P-gp, P-glycoprotein; MRP1,
multidrug resistance-associated protein 1; BCRP, breast cancer
resistance protein; MMP2, matrix metalloproteinase 2; p-FAK,
phosphorylated-focal adhesion kinase. |
Discussion
As a commonly expressed immunophenotypic marker of
B-ALL, CD9 is found to regulate the biological behavior of B-ALL
cells and to be associated with the clinical prognosis of B-ALL
patients (2–7). However, the corresponding mechanisms
underlying the effects of CD9 on the disease progression of B-ALL
remain to be explored. The PI3K/AKT signaling pathway is shown to
be involved in a variety of tumor-related biological functions, and
its role in tumorigenesis and development is also well-established
(8). It should be noted that
previously published experimental studies have demonstrated that
tetraspanin proteins are capable of regulating the PI3K/AKT
pathway. We therefore hypothesized that CD9 may be involved in the
regulation of the biological behavior of B-ALL cells through the
PI3K/AKT pathway. In order to investigate the relationship between
CD9 and the PI3K/AKT pathway, we determined the expression of
p-PI3K and p-AKT in B-ALL cell line SUP-B15 after CD9
knockdown. Results showed that p-PI3K expression and the p-AKT/AKT
ratio in the SUP-B15 cells was significantly reduced after
CD9 knockdown, suggesting that CD9 regulates the activity of
PI3K/AKT signaling pathway.
The PI3K/AKT pathway has been shown to be involved
in the regulation of cell proliferation and apoptosis via its
downstream cell cycle- and apoptosis-related molecules, such as
p53, p21 and cleaved caspase-3 (12–14).
Importantly, our previous study demonstrated that silencing of
CD9 upregulated the protein levels of p53, p21 and cleaved
caspase-3 in SUP-B15 cells, which may result in the suppression of
cell cycle progression and induction of apoptosis in SUP-B15 cells
(7). Additionally, the downregulation
of CD9 expression was found to increase the cytotoxicity of
chemotherapeutic agents in SUP-B15 cells (7). Although the drug-resistance of tumor
cells is quite complex, factors that may be involved, such as
ATP-binding cassette drug transporters P-gp, MRP1 and BCRP, have
been identified. More relatively, the activation of the PI3K/AKT
pathway has been reported to contribute to drug-resistance of tumor
cells through the upregulation of ATP-binding cassette drug
transporters (15). Similarly, the
present results indicated that the downregulation of CD9 expression
significantly reduced the protein levels of P-gp, MRP1 and BCRP in
SUP-B15 cells, suggesting that the CD9-regulated chemo-resistance
of B-ALL cells may be mediated by ATP-binding cassette drug
transporters, downstream targets of the PI3K/AKT pathway.
Metastasis and invasion are key clinicopathological
features of acute leukemia (16). Our
previous study demonstrated that downregulation of CD9 expression
inhibited cell adhesion, migration and invasion in SUP-B15 cells
(7). On the other hand, activation of
the PI3K/AKT pathway has been shown to promote adhesion, migration
and invasion of tumor cells by the upregulation of MMP2 and p-FAK
(17–19). The present study indicated that
CD9 knockdown significantly reduced MMP2 expression and the
p-FAK/FAK ratio in SUP-B15 cells, suggesting that the
downregulation of CD9 expression inhibited adhesion, migration and
invasion of B-ALL cells by suppressing the expression of MMP2 and
p-FAK through PI3K/AKT pathway.
PI3K, as a heterodimer composed of a p85-regulatory
subunit and a p110-catalytic subunit, could be recruited to
receptor tyrosine residues on the cell surface membrane and then be
activated by either phosphorylation or binding of adaptor proteins
to the Src Homlogy 2 domain of p85 subunit (20–25). As a
commonly mutated protein in solid tumors, PI3K-p85 plays a key role
in the activation of the PI3K/AKT pathway. Moreover, the
downregulation of PI3K-p85 expression has been shown to inhibit
proliferation and migration of tumor cells (26–29). We
therefore hypothesized that CD9 may activate the PI3K/AKT pathway
via the interaction with PI3K-p85. Two forms of PI3K-p85 have been
identified: PI3K-p85α is ubiquitously expressed and is encoded by
the Pik3r1 gene, whereas PI3K-p85β is also widely expressed
but is encoded by the Pik3r2 gene (30,31). In
the present study, GST pull-down assay showed the binding between
CD9 and both PI3K-p85α and PI3K-p85β in vitro, while
co-immunoprecipitation assay showed the binding between CD9 and
both PI3K-p85α and PI3K-p85β in vivo. These results further
indicated that CD9 could mediate proliferation, drug-resistance,
adhesion, migration and invasion of B-ALL cells through activation
of the PI3K/AKT pathway.
Furthermore, in order to assess the in vitro
anti-leukemia effects of the inhibition of the PI3K/AKT pathway in
B-ALL cells, LY294002 was used to culture SUP-B15 cells and thereby
manipulate the PI3K/AKT pathway. Meanwhile, we investigated the
effects of LY294002 treatment on the biological behavior as well as
PI3K/AKT pathway in SUP-B15 cells. The present results showed that
LY294002 treatment significantly inhibited cell proliferation,
adhesion, migration and invasion, while promoting the efficacy of
chemotherapeutic agents in SUP-B15 cells. We also found that
LY294002 treatment significantly reduced p-PI3K expression and the
p-AKT/AKT ratio, as well as drug-resistance-related (including
P-gp, MRP1 and BCRP) and motility-related factors (including MMP2
and p-FAK), while increasing the protein levels of cell cycle- and
apoptosis-related factors (including p53, p21 and cleaved
caspase-3). These findings suggest that inhibition of the PI3K/AKT
pathway may provide a novel treatment option for CD9+
B-ALL.
In conclusion, this study demonstrated that CD9
could modulate the biological behavior of B-ALL cells through the
PI3K/AKT singling pathway via direct interaction with PI3K-p85, and
the inhibition of PI3K/AKT pathway might be a novel treatment
option for CD9+ B-ALL. Further studies are needed to
verify the efficiency of the PI3K/AKT inhibitor in patient-derived
primary B-ALL cells and xenograft animal models for B-ALL.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This research was funded by the Natural Science
Foundation of Zhejiang Province (grant nos. LY21H080006 and
LQ19H080002), and the Public Welfare Science and Technology Project
of Wenzhou (grant no. Y20190119).
Availability of data and materials
The datasets generated or analyzed during this study
are not publicly available due to confidentiality of another study
from our group but are available from the corresponding authors
upon reasonable request.
Authors' contributions
JHF and KTL conceived of the research project. YFS,
ZYH, YSH, RJD, CYX and KY conducted the experiments and collected
the data. JHF, KTL and YHS analyzed the data. YFS and ZYH wrote the
paper. All authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hemler ME: Tetraspanin functions and
associated microdomains. Nat Rev Mol Cell Biol. 6:801–811. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liang P, Miao M, Liu Z, Wang H, Jiang W,
Ma S, Li C and Hu R: CD9 expression indicates a poor outcome in
acute lymphoblastic leukemia. Cancer Biomark. 21:781–786. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leung KT, Zhang C, Chan KYY, Li K, Cheung
JTK, Ng MHL, Zhang XB, Sit T, Lee WYW, Kang W, et al: CD9 blockade
suppresses disease progression of high-risk pediatric B-cell
precursor acute lymphoblastic leukemia and enhances
chemosensitivity. Leukemia. 34:709–720. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nishida H, Yamazaki H, Yamada T, Iwata S,
Dang NH, Inukai T, Sugita K, Ikeda Y and Morimoto C: CD9 correlates
with cancer stem cell potentials in human B-acute lymphoblastic
leukemia cells. Biochem Biophys Res Commun. 382:57–62. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yamazaki H, Xu CW, Naito M, Nishida H,
Okamoto T, Ghani FI, Iwata S, Inukai T, Sugita K and Morimoto C:
Regulation of cancer stem cell properties by CD9 in human B-acute
lymphoblastic leukemia. Biochem Biophys Res Commun. 409:14–21.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arnaud MP, Vallee A, Robert G, Bonneau J,
Leroy C, Varin-Blank N, Rio AG, Troadec MB, Galibert MD and
Gandemer V: CD9, a key actor in the dissemination of lymphoblastic
leukemia, modulating CXCR4-mediated migration via RAC1 signaling.
Blood. 126:1802–1812. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xing C, Xu W, Shi Y, Zhou B, Wu D, Liang
B, Zhou Y, Gao S and Feng J: CD9 knockdown suppresses cell
proliferation, adhesion, migration and invasion, while promoting
apoptosis and the efficacy of chemotherapeutic drugs and imatinib
in Ph+ ALL SUP-B15 cells. Mol Med Rep. 22:2791–2800.
2020.PubMed/NCBI
|
|
8
|
Fruman DA, Chiu H, Hopkins BD, Bagrodia S,
Cantley LC and Abraham RT: The PI3K pathway in human disease. Cell.
170:605–635. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sugiura T and Berditchevski F: Function of
alpha3beta1-tetraspanin protein complexes in tumor cell invasion.
Evidence for the role of the complexes in production of matrix
metalloproteinase 2 (MMP-2). J Cell Biol. 146:1375–1389. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sawada S, Yoshimoto M, Odintsova E,
Hotchin NA and Berditchevski F: The tetraspanin CD151 functions as
a negative regulator in the adhesion-dependent activation of Ras. J
Biol Chem. 278:26323–26326. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qi JC, Wang J, Mandadi S, Tanaka K,
Roufogalis BD, Madigan MC, Lai K, Yan F, Chong BH, Stevens RL and
Krilis SA: Human and mouse mast cells use the tetraspanin CD9 as an
alternate interleukin-16 receptor. Blood. 107:135–142. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang L, Zhu S, Shi X and Sha W: The
silence of p66(Shc) in HCT8 cells inhibits the viability via
PI3K/AKT/Mdm-2/p53 signaling pathway. Int J Clin Exp Pathol.
8:9097–9104. 2015.PubMed/NCBI
|
|
13
|
Tang C, Lu YH, Xie JH, Wang F, Zou JN,
Yang JS, Xing YY and Xi T: Downregulation of survivin and
activation of caspase-3 through the PI3K/Akt pathway in ursolic
acid-induced HepG2 cell apoptosis. Anticancer Drugs. 20:249–258.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang H, Jia XH, Chen JR, Yi YJ, Wang JY,
Li YJ and Xie SY: HOXB4 knockdown reverses multidrug resistance of
human myelogenous leukemia K562/ADM cells by downregulating P-gp,
MRP1 and BCRP expression via PI3K/Akt signaling pathway. Int J
Oncol. 49:2529–2537. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Klein G, Vellenga E, Fraaije MW, Kamps WA
and de Bont ES: The possible role of matrix metalloproteinase
(MMP)-2 and MMP-9 in cancer, e.g. acute leukemia. Crit Rev Oncol
Hematol. 50:87–100. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou R, Xu L, Ye M, Liao M, Du H and Chen
H: Formononetin inhibits migration and invasion of MDA-MB-231 and
4T1 breast cancer cells by suppressing MMP-2 and MMP-9 through
PI3K/AKT signaling pathways. Horm Metab Res. 46:753–760. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yadav V and Denning MF: Fyn is induced by
Ras/PI3K/Akt signaling and is required for enhanced
invasion/migration. Mol Carcinog. 50:346–352. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tai YL, Chen LC and Shen TL: Emerging
roles of focal adhesion kinase in cancer. Biomed Res Int.
2015:6906902015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Di Zazzo E, Feola A, Zuchegna C, Romano A,
Donini CF, Bartollino S, Costagliola C, Frunzio R, Laccetti P, Di
Domenico M and Porcellini A: The p85 regulatory subunit of PI3K
mediates cAMP-PKA and insulin biological effects on MCF-7 cell
growth and motility. ScientificWorldJournal. 2014:5658392014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Breitkopf SB, Yang X, Begley MJ, Kulkarni
M, Chiu YH, Turke AB, Lauriol J, Yuan M, Qi J, Engelman JA, et al:
A cross-species study of pi3k protein-protein interactions reveals
the direct interaction of P85 and SHP2. Sci Rep. 6:204712016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee JY, Chiu YH, Asara J and Cantley LC:
Inhibition of PI3K binding to activators by serine phosphorylation
of PI3K regulatory subunit p85alpha Src homology-2 domains. Proc
Natl Acad Sci USA. 108:14157–14162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Comb WC, Hutti JE, Cogswell P, Cantley LC
and Baldwin AS: p85α SH2 domain phosphorylation by IKK promotes
feedback inhibition of PI3K and Akt in response to cellular
starvation. Mol Cell. 45:719–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hofmann BT and Jücker M: Activation of
PI3K/Akt signaling by n-terminal SH2 domain mutants of the p85α
regulatory subunit of PI3K is enhanced by deletion of its
c-terminal SH2 domain. Cell Signal. 24:1950–1954. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jaiswal BS, Janakiraman V, Kljavin NM,
Chaudhuri S, Stern HM, Wang W, Kan Z, Dbouk HA, Peters BA, Waring
P, et al: Somatic mutations in p85alpha promote tumorigenesis
through class IA PI3K activation. Cancer Cell. 16:463–474. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Folgiero V, Di Carlo SE, Bon G, Spugnini
EP, Di Benedetto A, Germoni S, Pia Gentileschi M, Accardo A,
Milella M, Morelli G, et al: Inhibition of p85, the non-catalytic
subunit of phosphatidylinositol 3-kinase, exerts potent antitumor
activity in human breast cancer cells. Cell Death Dis. 3:e4402012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun M, Hillmann P, Hofmann BT, Hart JR and
Vogt PK: Cancer-derived mutations in the regulatory subunit
p85alpha of phosphoinositide 3-kinase function through the
catalytic subunit p110alpha. Proc Natl Acad Sci USA.
107:15547–15552. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Feola A, Cimini A, Migliucci F, Iorio R,
Zuchegna C, Rothenberger R, Cito L, Porcellini A, Unteregger G,
Tombolini V, et al: The inhibition of p85αPI3KSer83 phosphorylation
prevents cell proliferation and invasion in prostate cancer cells.
J Cell Biochem. 114:2114–2119. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fruman DA, Snapper SB, Yballe CM, Davidson
L, Yu JY, Alt FW and Cantley LC: Impaired B cell development and
proliferation in absence of phosphoinositide 3-kinase p85alpha.
Science. 283:393–397. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Terauchi Y, Tsuji Y, Satoh S, Minoura H,
Murakami K, Okuno A, Inukai K, Asano T, Kaburagi Y, Ueki K, et al:
Increased insulin sensitivity and hypoglycaemia in mice lacking the
p85 alpha subunit of phosphoinositide 3-kinase. Nat Genet.
21:230–235. 1999. View
Article : Google Scholar : PubMed/NCBI
|