Introduction
In a worldwide perspective, kidney cancer, one of
the most commonly diagnosed malignant tumor, accounts for
approximately 2–3% of all types of cancers (1–3). Clear
cell renal cell carcinoma (RCC) is considered to be the main type
of RCC, and its incidence and morbidity is consistently increasing
annually (4). In the clinic, the
prognosis of RCC patients may be favorable if diagnosed at an early
stage. RCC is an intricate disease owing to its etiological and
histological heterogeneity, and the underlying molecular mechanisms
of RCC tumorigenesis are far from clear (5,6). Exploring
the concrete mechanisms of RCC and identifying various potential
biomarkers for the early diagnosis of RCC are crucial.
Currently, with the rapid development of
quantitative proteomic methods by tandem mass spectrometry (MS/MS),
researchers are able to explore the total differentially expressed
proteins in different cancers and certain post-translational
modifications (PTMs) of crucial proteins in cancers, which is
helpful for screening protein candidates serving as novel
diagnostic biomarkers or therapeutic targets (7–9).
PTMs of protein have been demonstrated to have a
major influence in the regulation of protein functions. There are
multiple modifications including phosphorylation, acetylation,
SUMOylation, succinylation and malonylation (10,11). Among
these PTMs, phosphorylation is fast, reversible and often highly
specific, which remains a critical protein modification for
regulating signaling pathways in various biological processes
(12). Activation of protein
phosphorylation contributes to a cascade of downstream signaling
processes which are critical for cell maintenance and survival, and
disorders of this process have been implicated in various types of
cancer (13,14). Thus, the comprehensive identification
of phosphorylation is pivotal by which to explore the molecular
pathogenesis, which may contribute to the precise treatment and
prediction of RCC. To date, various studies have specifically
focused on the changes in the quantitative global proteome but few
have investigated the phosphorylation in RCC (15–17).
Therefore, we aimed to combine quantitative global proteome
analysis and alterations in phosphorylation, with the expectation
to deepen insight into the tumorigenesis of RCC.
Materials and methods
RCC sample collection
Nine human RCC tumor samples and matched adjacent
non-tumor samples were collected from RCC patients treated at the
Urology Department of Sir Run Run Shaw Hospital from February to
December, 2018 (Zhejiang, China). Inclusion criteria included: i)
samples from patients with RCC stored in the biological specimen
bank of Sir Run Run Shaw Hospital; ii) patients aged ≥18 years; and
iii) cancer type identified histologically or cytologically.
Exclusion criteria included: i) patients with severe organic
lesions; ii) patients with additional complications such as renal
cyst, kidney stone and other kidney diseases; iii) patients
previously diagnosed with any other malignancies. Four RCC tumor
samples and matched adjacent non-tumor samples were used for liquid
chromatography-mass spectrometry (LC/MS) and five paired samples
were used for immunohistochemistry (IHC). The patients included 5
males and 4 females, with an average age of 53.6±7.6 years (range,
43–60 years). Patient information is documented in Table SI. The signed and dated consent form
of each patient was obtained before surgery. All samples used in
this research were stored at the biological specimen bank of Sir
Run Run Shaw Hospital. These samples were obtained with the
permission of the Ethics Committee of Sir Run Run Shaw Hospital
affiliated with Zhejiang University Medical College. None of the
RCC patents received any other therapy before surgery. Human tumor
samples were treated with phosphate-buffered saline (PBS) three
times to be cleansed from the residual blood, and then maintained
in liquid nitrogen and finally stored at −80°C.
Protein extraction
Firstly, we took out the sample from −80°C, weighed
a suitable amount of tissue sample into the mortar and added liquid
nitrogen and fully ground them to powder. The samples of each group
were respectively added with 4 times the volume of the powder lysis
buffer (8 M urea, 1% protease inhibitor, 1% phosphatase inhibitor)
followed by sonication three times on ice. At 4°C, centrifugation
at 12,000 × g was used for 10 min. The cell fragments were removed,
the supernatant was transferred to a new centrifuge tube, and the
protein concentration was determined using a BCA kit (Abcam).
Trypsin digestion
In preparation of trypsin digestion, the final
concentration of dithiothreitol was 5 mM and temperature was
reduced to 56°C for 30 min. After that, the final concentration of
iodoacetamide was 11 mM and incubated with the protein sample in
darkness for 15 min at room temperature. Finally, the urea
concentration of the sample was diluted to less than 2 M. Trypsin
was added in a mass ratio of 1:50 (pancreatin:protein) and
digestion was performed overnight at 37°C. The trypsin was added at
the ratio of 1:100 (pancreatin:protein) and digestion was continued
for 4 h.
Tandem mass tag (TMT) labeling
The peptides were desalted using Strata X C18 column
(Phenomenex) and freeze-dried in a vacuum. The peptide powder was
dissolved with 0.5 M tetraethylammonium bromide (TEAB) and labeled
according to the operating instructions of the TMT kit (Thermo
Fisher Scientific, Inc.). The simple operation process was as
follows: after unfreezing, the labeling reagent was dissolved with
acetonitrile, mixed with peptide sample, incubated at room
temperature for 2 h, and pooled, desalted, and dried by vacuum
centrifugation.
High-performance liquid chromatography
(HPLC) fractionation
The peptide sample was classified by high pH reverse
HPLC using a Thermo Scientific™ BetaSil™ C18 HPLC Column (Thermo
Fisher Scientific, Inc.) (5-µm particle size, 10-mm internal
diameter, 250-mm length). The simple operation process was as
follows: 60 fractions were separated for 60 min with the peptide
grading gradient of 8–32% acetonitrile and pH of 9.0. Next, for the
proteomics analysis, the peptide fractions were combined into 18
fractions, and for the phosphorylation proteomics analysis, the
peptide was combined into 8 fractions, and the combined components
were dried by vacuum centrifugation.
Affinity enrichment
For phosphorylated peptide enrichment, the peptide
sample was dissolved in enrichment buffer solution (50%
acetonitrile/6% trifluoroacetic acid). The supernatant was then
transferred to a new centrifuge tube and 30 mg IMAC materials was
added. The mixed sample was placed on a rotating shaker and
incubated gently. After incubation, the resin was washed three
times with wash buffer solution of 50% acetonitrile/6%
trifluoroacetic acid and 30% acetonitrile/0.1% trifluoroacetic
acid. Finally, 10% ammonia buffer was used to elute the
phosphopeptide, and the eluent was collected and vacuum
freeze-dried. Desalting was carried out according to the
instructions of the C18 ziptips after drying, and analysis was
followed by liquid chromatography-tandem mass spectrometry
(LC-MS/MS).
Immunohistochemical staining
Samples were fixed in 10% (v/v) formaldehyde in PBS,
embedded in paraffin and cut into 4-µm sections and used for IHC
staining with antibodies. To enhance antigen exposure, the slides
were treated with 1X EDTA at 98°C for 10 min for antigen retrieval.
The slides were incubated with endogenous peroxidase blocking
solution to inhibit endogenous peroxidase and were then incubated
with the primary antibody (CD14, ab183322; Abcam, dilution 1:100;
MPO, ab208670; Abcam, dilution 1:1,000; NCF2, Abs135832, Absin,
dilution 1:100; SOD2, ab246860; Abcam, dilution 1:1,000; PARP1,
ab194586; Abcam, dilution 1:50; MUT, ab240091; Abcam, dilution
1:200; ACADM, ab239914; Abcam, dilution 1:500; PCK1, ab248573;
Abcam, dilution 1:200) at room temperature for 60 min. After
rinsing with Tris-buffered saline, the slides were incubated for 45
min with biotin-conjugated secondary antibody (catalog no.
SA00004-2; Proteintech Wuhan Sanying, dilution 1:600), washed, and
then incubated with enzyme conjugate HRP-streptavidin. Freshly
prepared DAB (Zymed; Thermo Fisher Scientific, Inc.) was used as a
substrate to detect HRP. Finally, slides were counterstained with
hematoxylin and mounted with aqueous mounting media. Studies on
human tissue samples were conducted with approval from the Ethics
Committee of the Sir Run Run Shaw Hospital, Zhejiang
University.
Bioinformatic analyses
Excel was used for investigation of the
differentially expressed proteins with quantification ratios
>1.5 as the upregulation threshold and <0.67 as the
downregulation threshold. A P-value <0.05 was considered
statistically significant. Database for Annotation, Visualization
and Integrated Discovery (DAVID, http://david.ncifcrf.gov) was performed to classify
proteins into three categories (biological process, cellular
compartment, and molecular function) against the background of
Homo sapiens. Online Search Tool for the Retrieval of
Interacting Genes (STRING) database (http://www.string-db.org) was performed to evaluate
the protein-protein interaction (PPI) information. We imported the
differentially expressed proteins (DEPs) to STRING, and only
experimentally validated interactions with a combined scored
>0.7 were selected as significant. Then these significant DEPs
were mapped into Cytoscape plugin (https://cytoscape.org) to create network
visualizations. Finally, we put the resulting PPI network to module
analysis with the Plugin MCODE with the default parameters (Degree
cutoff ≥2, Node score cutoff ≥2, K-core ≥2, and Max depth=100).
Moreover, function and pathway enrichment analysis were performed
for differentially expressed genes (DEGs) in the modules. The
patient prognosis in regards to these proteins was analyzed by
online software GEPIA (http://gepia.cancer-pku.cn/index.html). We used Kyoto
Encyclopedia of Genes and Genomes (KEGG, http://www.kegg.jp) database to identify enriched
pathways. P<0.05 was considered as indicative of a statistical
significant result. All experiments described in this study were
performed during 2018 to 2019.
Results
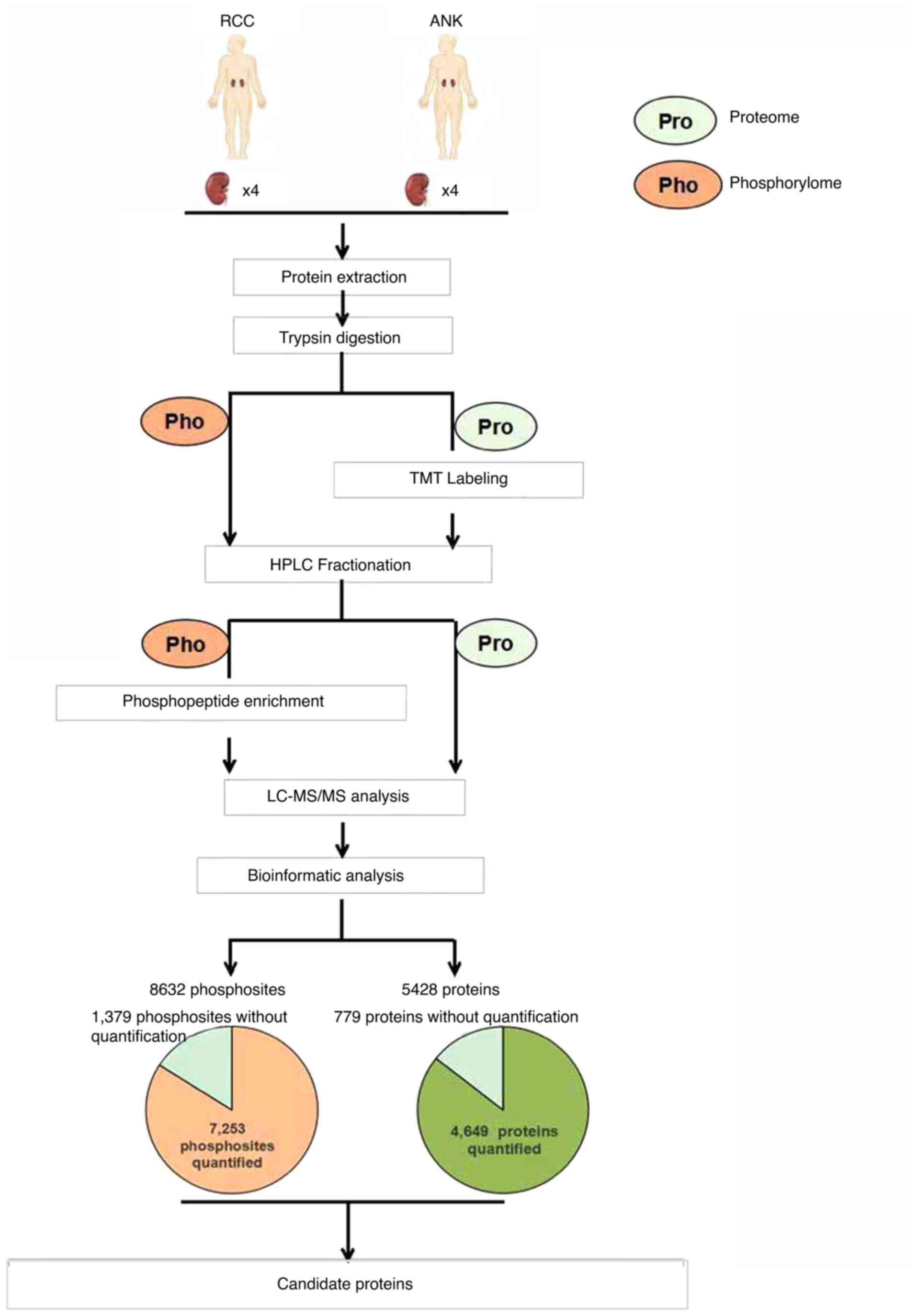
Workflow for quantitative proteome and
phosphorylome in RCC
Protein samples from 4 paired renal cell carcinoma
(RCC) and matched adjacent normal kidney (ANK) tissues were
harvested and analyzed for protein and phosphosite identification
using LC-MS/MS. TMT labeling and high-resolution mass spectrometry
(HRMS) were applied to ascertain the differences in the proteome
and phosphorylation between tumor and ANK tissues. Bioinformatic
analysis was used to conduct detailed studies in ccRCC. The
workflow is briefly indicted in Fig.
1.
Functional analysis of the global
proteome profiling in RCC
To identify the whole proteome profiling in RCC, we
employed TMT labeling and HPLC fractionation after high-resolution
LC-MS/MS analysis. Overall, 5,428 proteins were identified, and
4,649 of these proteins were quantified from four pairs of RCC
tissue samples. Using quantification ratios >1.5 as the
upregulation threshold and <0.67 as the downregulation
threshold, a P-value <0.05 was considered to indicate
statistical significance when comparing tumor samples to the
matched adjacent nontumor tissues. In total, 331 proteins were
upregulated and 378 proteins were downregulated.
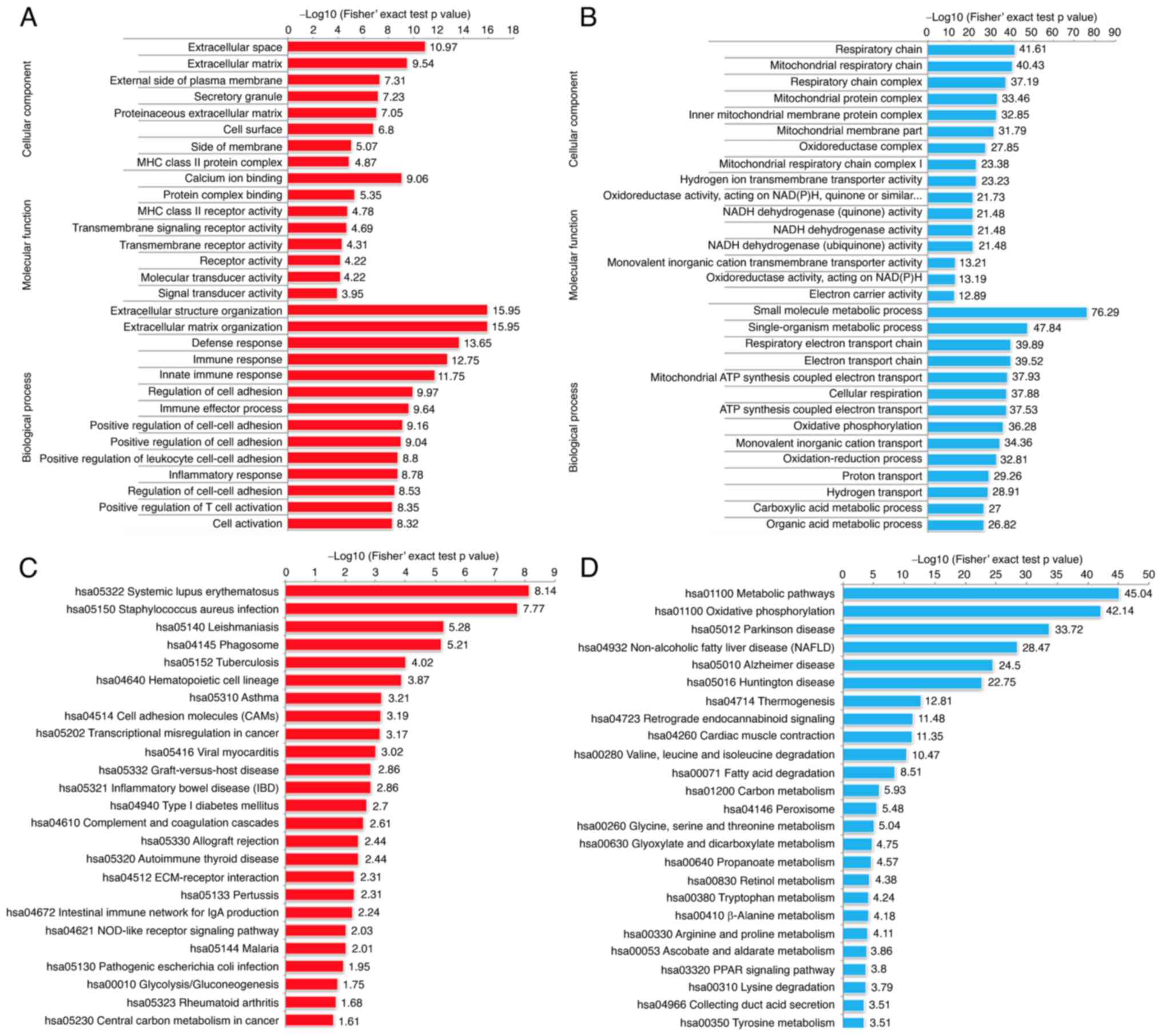
In order to further characterize the molecular
functions of these DEPs, we performed Gene Ontology (GO) enrichment
analysis (Fig. 2A and B). In terms of
biological process, the RCC tissues possessed significantly altered
biological metabolic processes, such as extracellular structure
organization, immune response, and regulation of cell adhesion.
Among the cellular component category, the RCC relative upregulated
proteins were mainly localized in the extracellular space,
extracellular matrix and cell surface, while the downregulated
proteins were mainly enriched in the respiratory chain. In terms of
molecular function, we observed that the upregulated proteins
exhibited significant enrichment in calcium ion binding, receptor
activity and signal transducer activity, while the downregulated
proteins were enriched in NADH dehydrogenase activity.
The KEGG pathway enrichment analysis was used to
explore the biological functions of these significantly
dysregulated proteins. The results of the KEGG pathway enrichment
indicated that the upregulated proteins exhibited functions
including cell adhesion molecules (CAMs), phagosome and
transcriptional dysregulation in cancer (Fig. 2C and D). Interestingly, we also
identified some immune-related genes, such as histocompatibility-1
MHC (MHCI), histocompatibility-2 MHC (MHCII), CD14
molecule (CD14), myeloperoxidase (MPO), protein
tyrosine phosphatase, receptor type, C (PTPRC) and integrin
subunit β2 (ITGB2), which indicated that immune response may
play pivotal roles in the tumor development of RCC. Consistent with
the Gene Ontology (GO) analysis, the result of KEGG pathway
enrichment analysis presented that the RCC tissues possessed
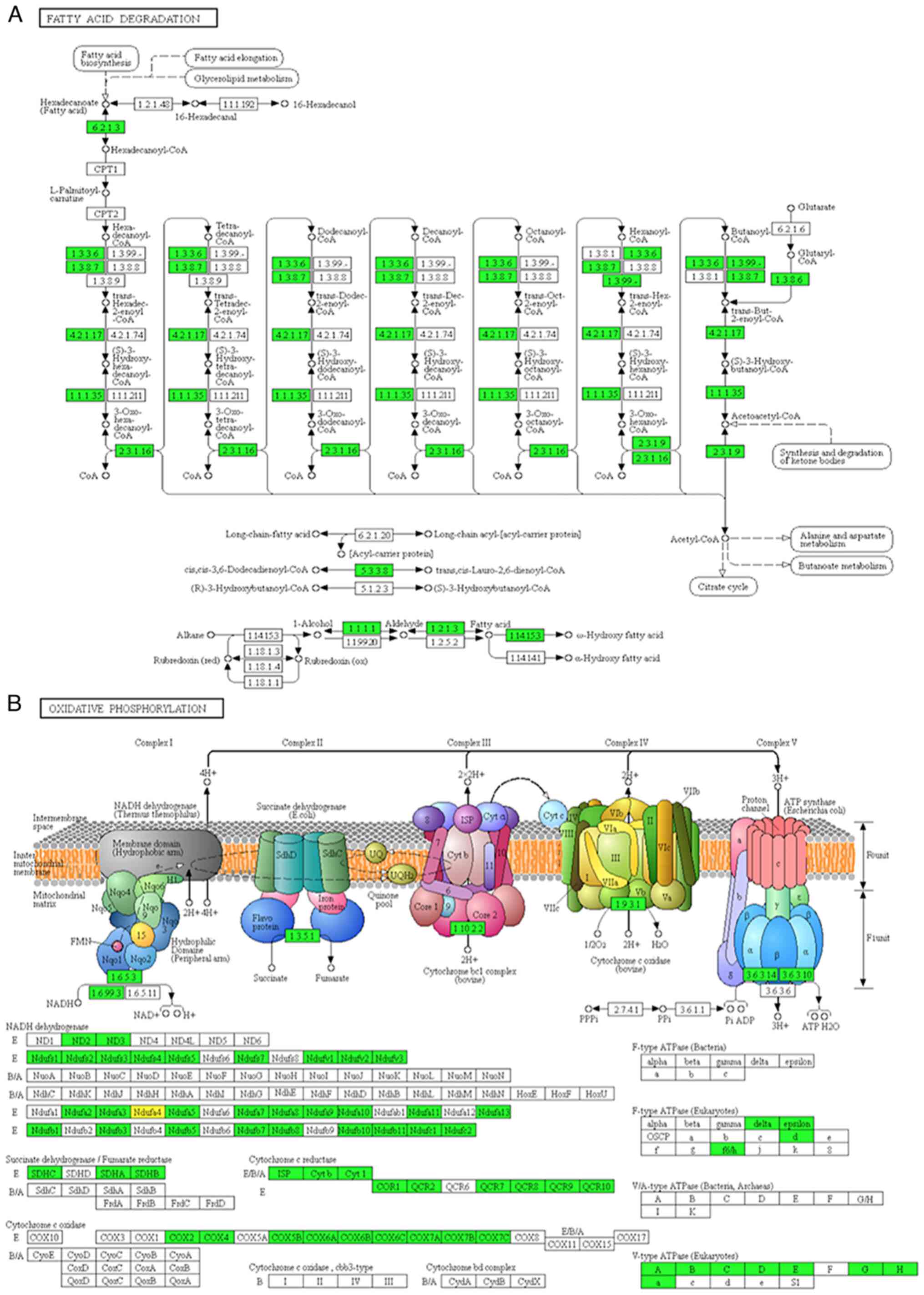
significantly altered biological metabolism, for example, fatty
acid metabolism and oxidative phosphorylation. The major proteins
and key enzymes related to fatty acid degradation and oxidative
phosphorylation were clearly decreased (Fig. 3A and B). Consistent with previous
studies (2,3), our results also indicated that a hypoxic
microenvironment is a typical characteristic during RCC
tumorigenesis.
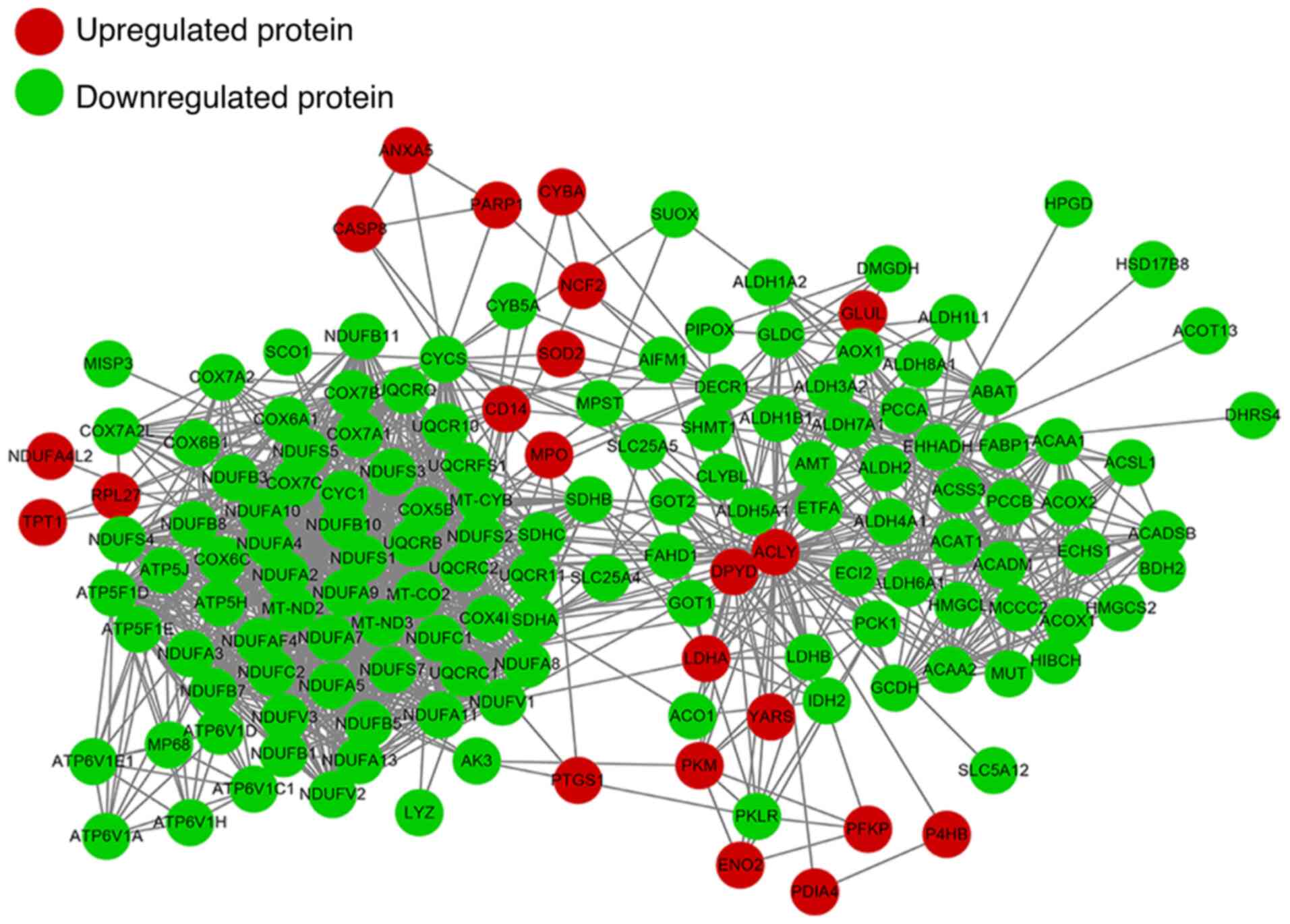
Protein-protein interactome
networks
We constructed a PPI network to present the main
interactions and regulatory relationships of all of these proteins
in RCC (Fig. 4). Expression of
lactate dehydrogenase A (LDHA3), vimentin 3, nicotinamide
N-methyltransferase (NNMT2), Annexin A43, larval cuticular protein,
14 kDa (LCP14) and enolase 21 (ENO21) in RCC tissue have been shown
to be dramatically higher in tumors when compared to the normal
tissue. In this study we ascertained that the above genes were
highly expressed in RCC tissues when compared to that in the normal
tissues. In addition, we found several new candidate proteins such
as CD14 molecule (CD14), myeloperoxidase (MPO), neutrophil
cytosolic factor 2 (NCF2), superoxide dismutase 2 (SOD2),
poly(ADP-ribose) polymerase 1 (PARP1), phosphofructokinase,
platelet α (PFKPA) that were upregulated and methylmalonyl-CoA
mutase (MUT), acyl-CoA dehydrogenase medium chain (ACADM),
phosphoenolpyruvate carboxykinase 1 (PCK1) that were downregulated
in RCC, which may be recognized as new biomarkers for RCC.
Phosphorylation profile of RCC and the
matched ANK tissues
Quantitative protein phosphorylation analyses were
performed by phosphopeptide enrichment using Ti-IMAC microspheres
(J&K Scientific) and the following LC-MS/MS analysis. All the
protein phosphorylation sites obtained from Maxquant were filtered
based on the localization probability, and setting the localization
probability >0.75 as the threshold. In general, of the
identified 8,632 phosphorylation sites in 3,128 protein groups in
the human tissues, 7,253 of these sites in 2,962 proteins were
quantified. In total, 315 differentially expressed phosphorylation
sites were quantified as downregulated targets and 334 sites were
quantified as upregulated targets with a quantification ratio
>1.5 for the upregulation threshold and <0.67 for the
downregulation threshold (P<0.05).
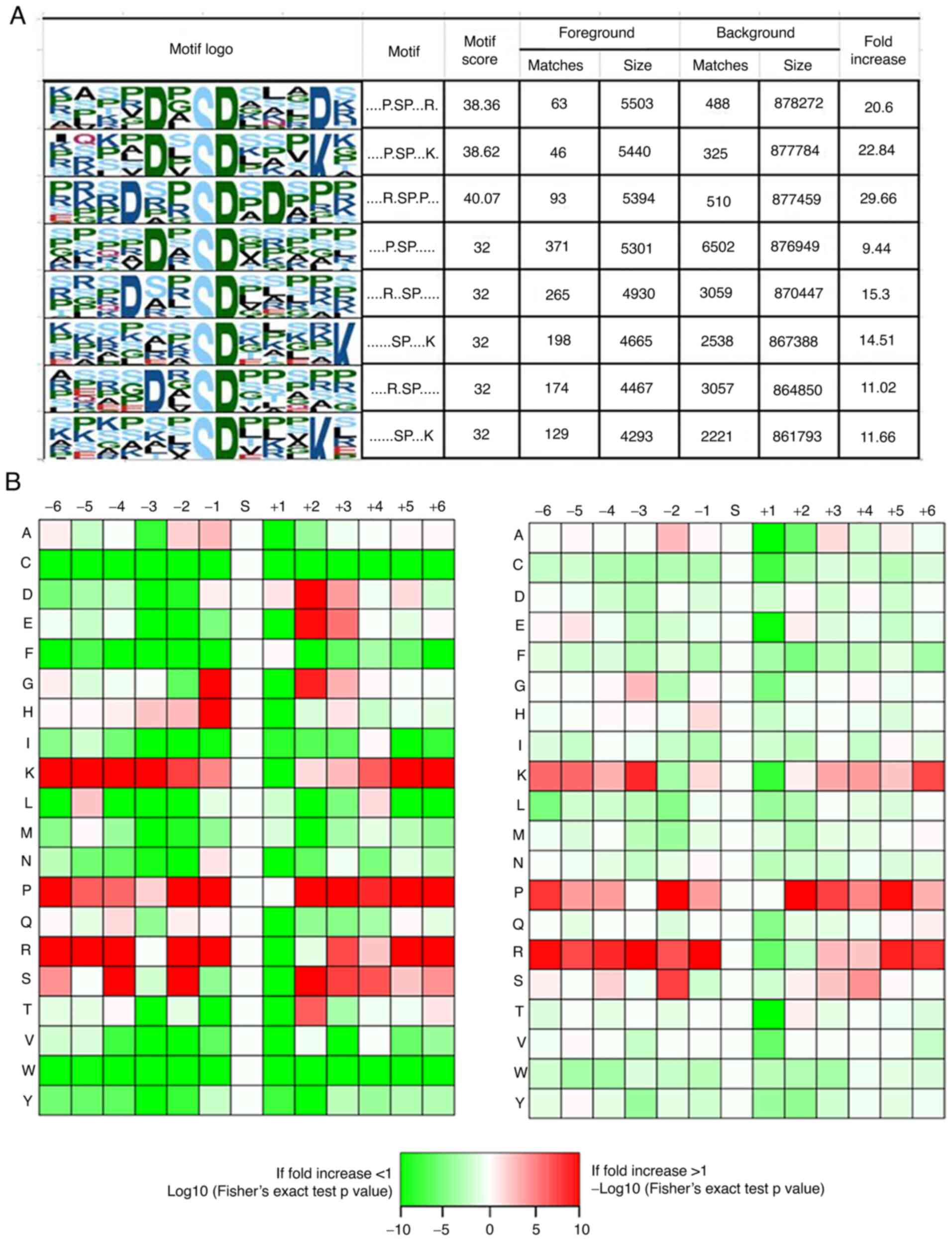
In order to determine the possible specific sequence
motifs surrounding the phosphorylated residues in the human RCC
samples, we carried out motif analysis to observe the amino acids
at the positions around the phosphorylation sites. Eight remarkably
enriched motifs were identified among all the identified
phosphorylation sites, namely, P*SP***R, P*SP***K, R**SP*P, P*SP,
R**SP, SP****K, R*SP and SP***K (where * represents a random amino
acid residue, Fig. 5A). The amino
acid frequencies of the sequences surrounding the phosphorylation
sites were analyzed to confirm whether the amino acids occurred
adjacent to phosphorylation sites are position-specific in motifs
(Fig. 5B). We identified that lysine
(K), proline (P) and arginine (R) were overrepresented in multiple
positions (±6, ±5, ±4, +3, −2, −1) surrounding the phosphorylation
sites, while cysteine (C), and tryptophane (W) were usually
depleted at all positions (±6, ±5, ±4, ±3, ±2, ±1). Of interest,
both glycine (G) and histidine (H) occurred more frequently at the
−1 position but few at the +1 position surrounding the
phosphorylation sites.
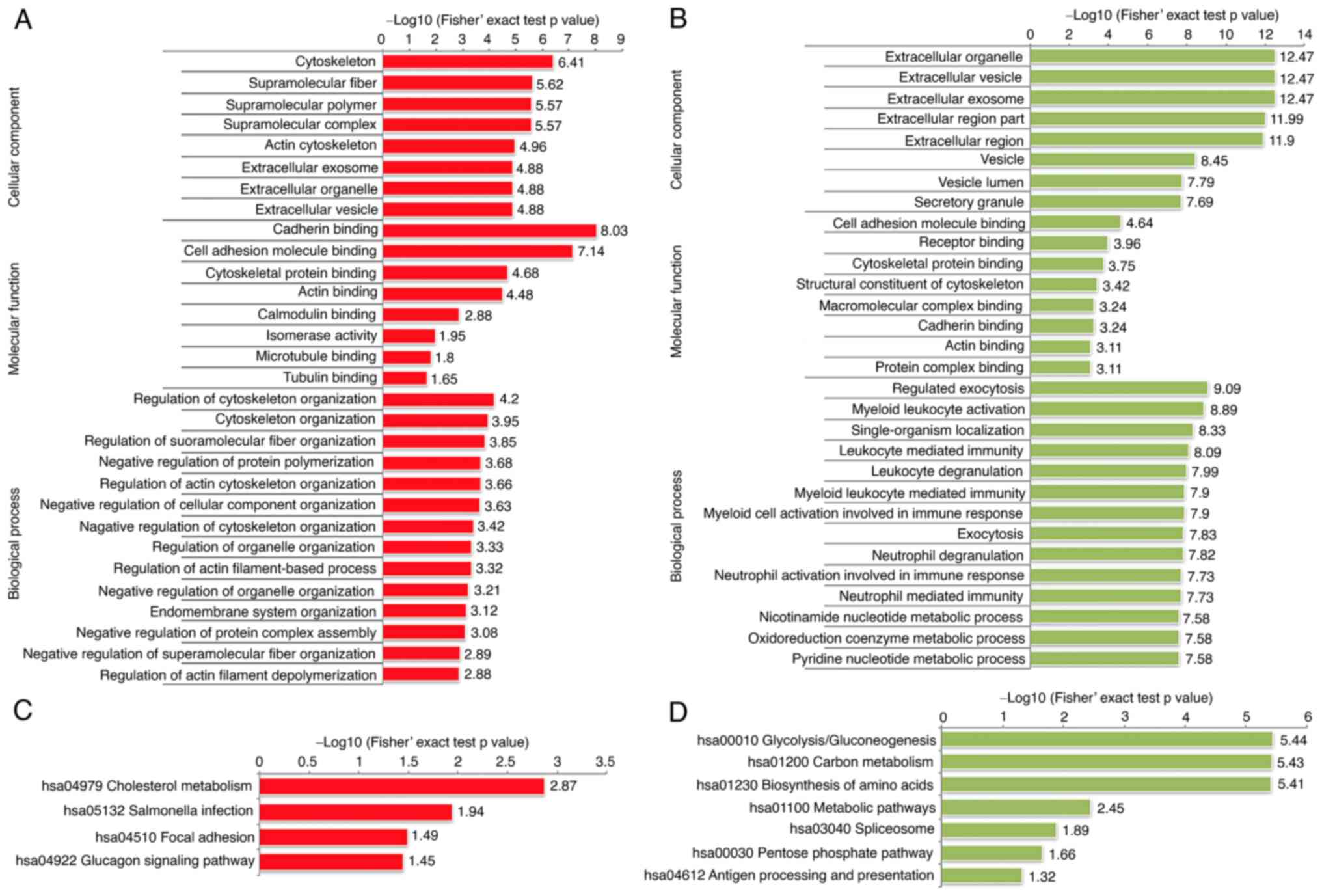
Bioinformatic analysis of the protein
phosphorylation in RCC
To evaluate the biological characteristics and
functional alterations of the phosphorylation in RCC tissues, GO
enrichment and KEGG analysis were performed. In terms of cellular
components, the dysregulated phosphorylated proteins were mainly
localized in the cytoskeleton, supramolecular complex, and
extracellular region. The principal molecular functions of these
proteins were mainly enriched in cytoskeletal protein binding and
cell adhesion molecule binding. As to the biological process, the
results indicated that the proteins related to cytoskeleton
organization, supramolecular fiber organization and regulation of
cellular component organization were relatively enriched among the
upregulated proteins; however, most of the downregulated proteins
were enriched in metabolic process and immune response (Fig. 6A and B). Interestingly, the KEGG
pathway analyses presented that the upregulated phosphorylated
proteins were also significantly enriched in focal adhesion. In
contrast, the enrichment pathways among the downregulated proteins
were glycolysis, biosynthesis of amino acids, and carbon metabolism
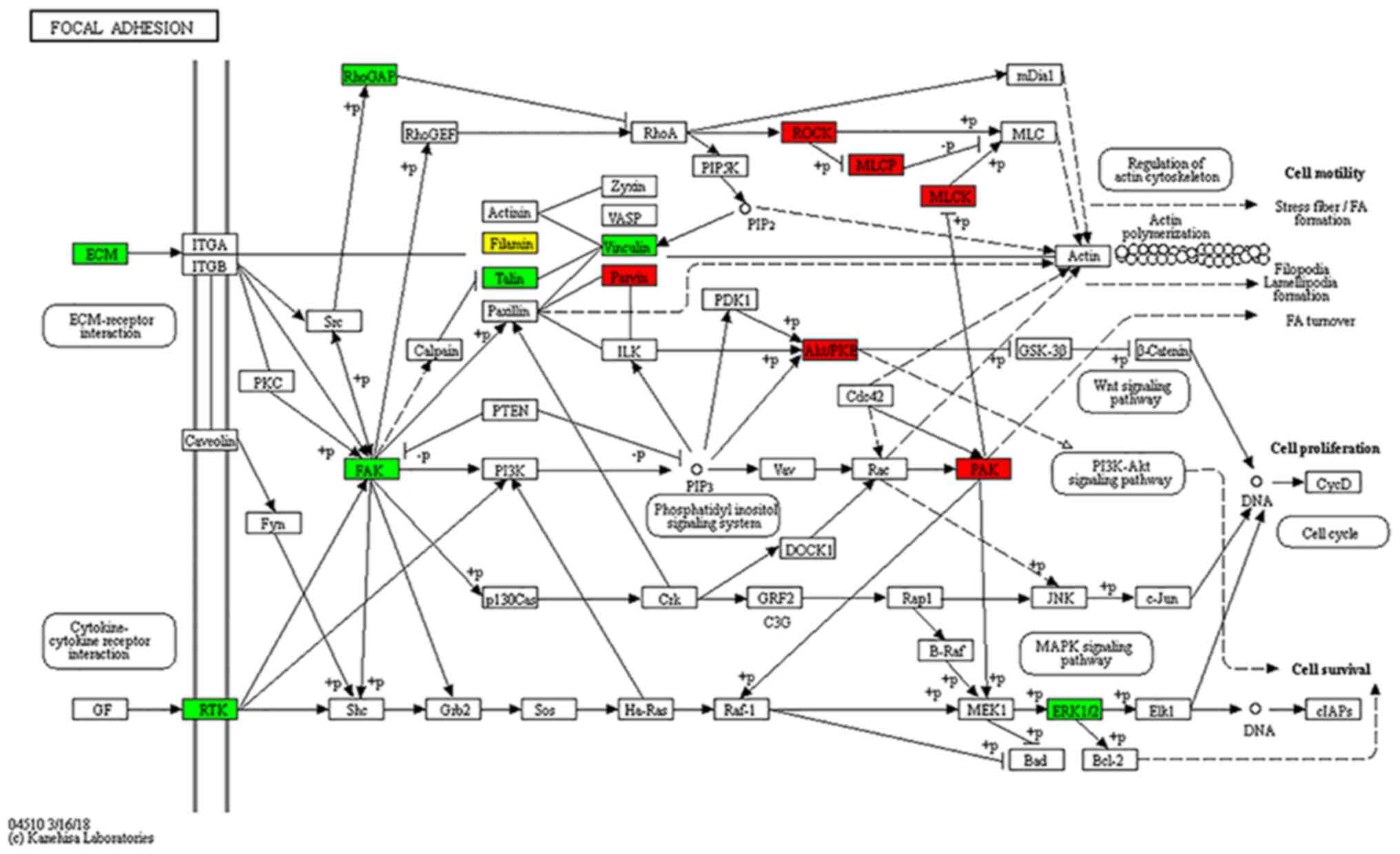
(Fig. 6C and D). Differential
phosphorylated proteins in focal adhesion pathways related to
cancer metastasis (cell motility, cell proliferation and cell
survival) are shown in Fig. 7. The
upregulated phosphorylated proteins were found to be closely
related to regulation of cellular cytoskeleton and significant
signaling pathways, which may probably contribute to metastasis of
RCC.
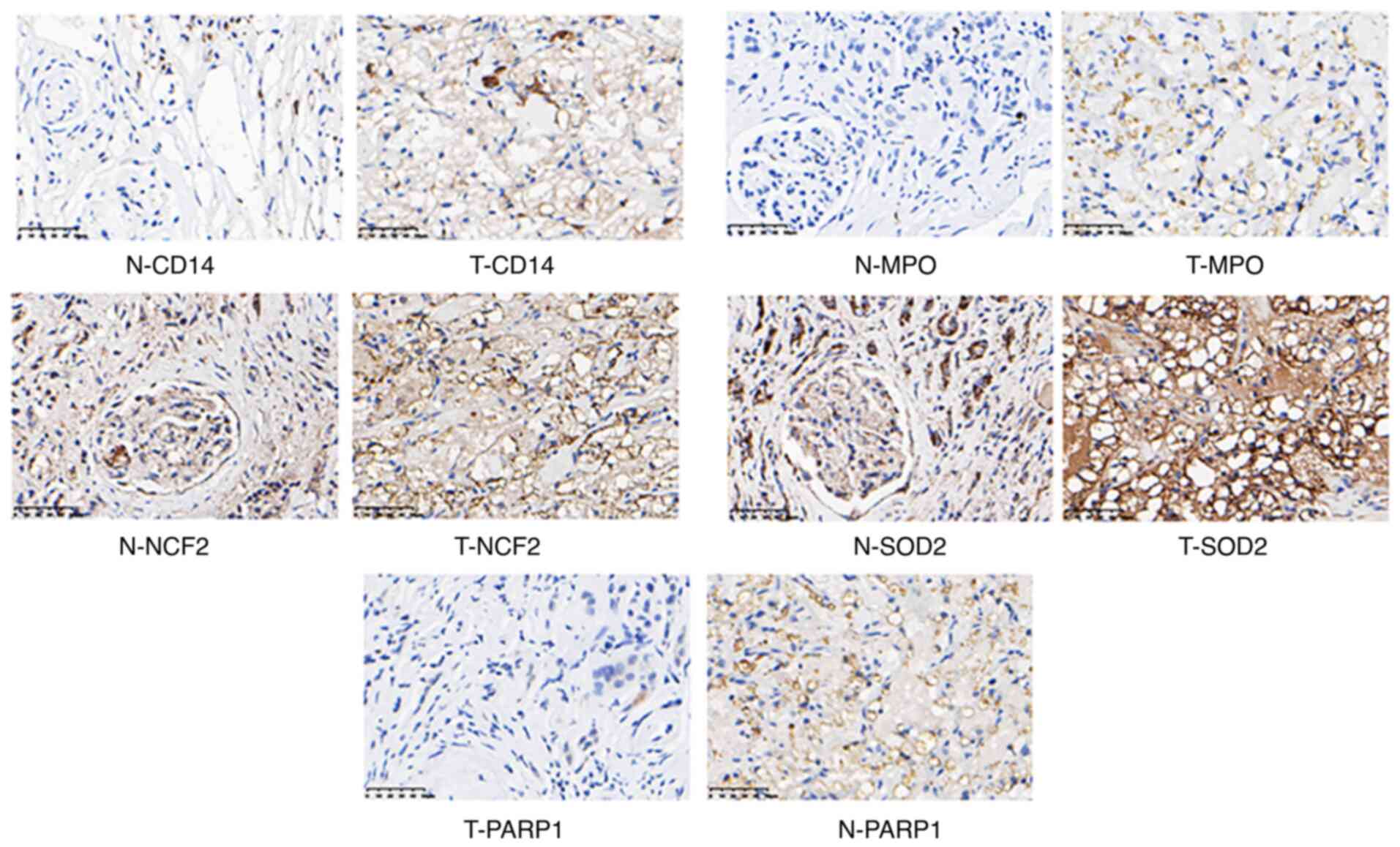
Verification of candidate proteins and
analysis of prognosis
To demonstrate the expression of candidate proteins
identified by the above analysis, IHC experiments were performed
using 5 human RCC tumor samples and matched adjacent non-tumor
samples. CD14, MPO, NCF2, SOD2, and PARP1 were upregulated
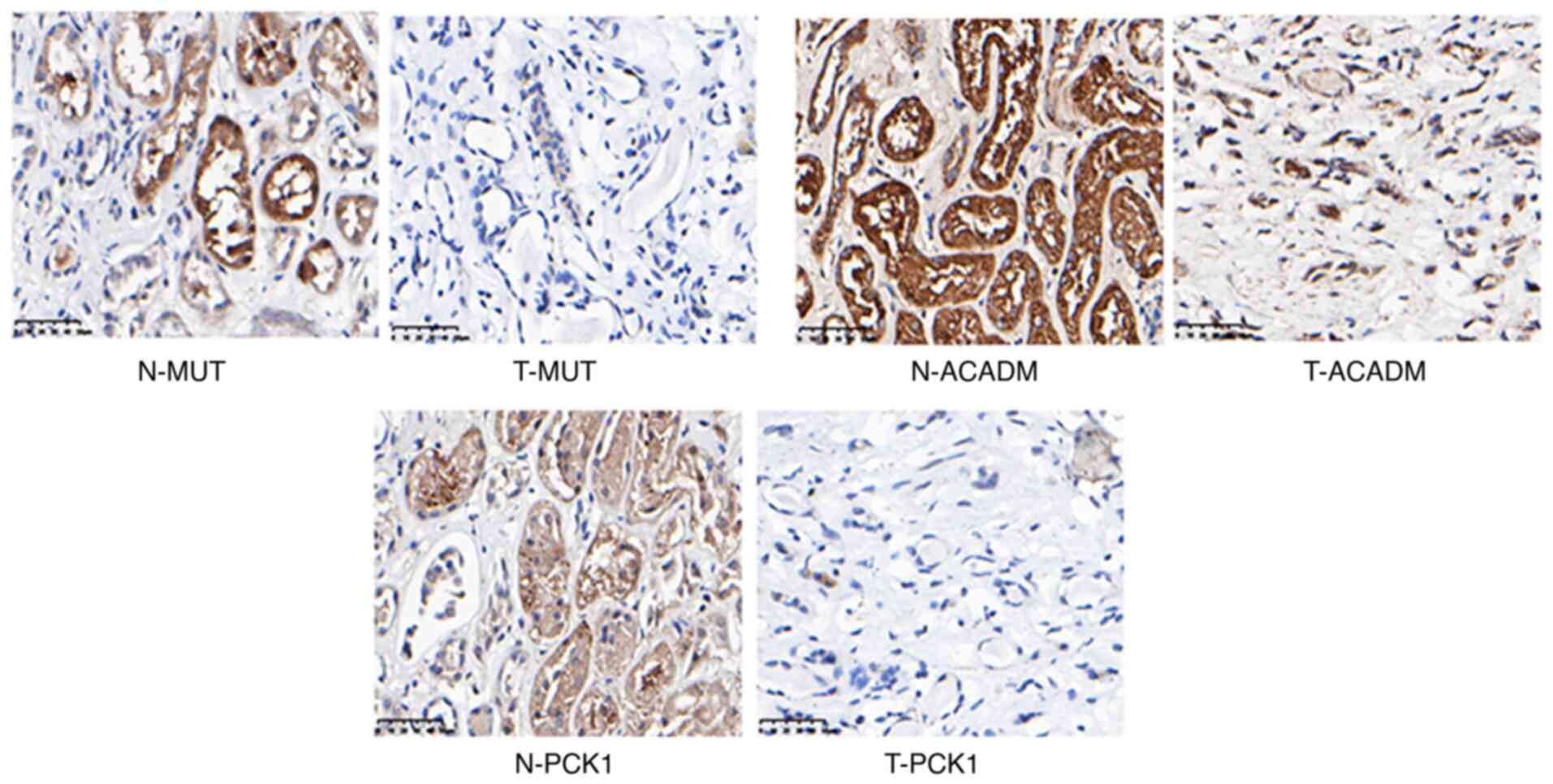
(Fig. 8) and MUT, ACADM and PCK1 were
downregulated in RCC (Fig. 9). In
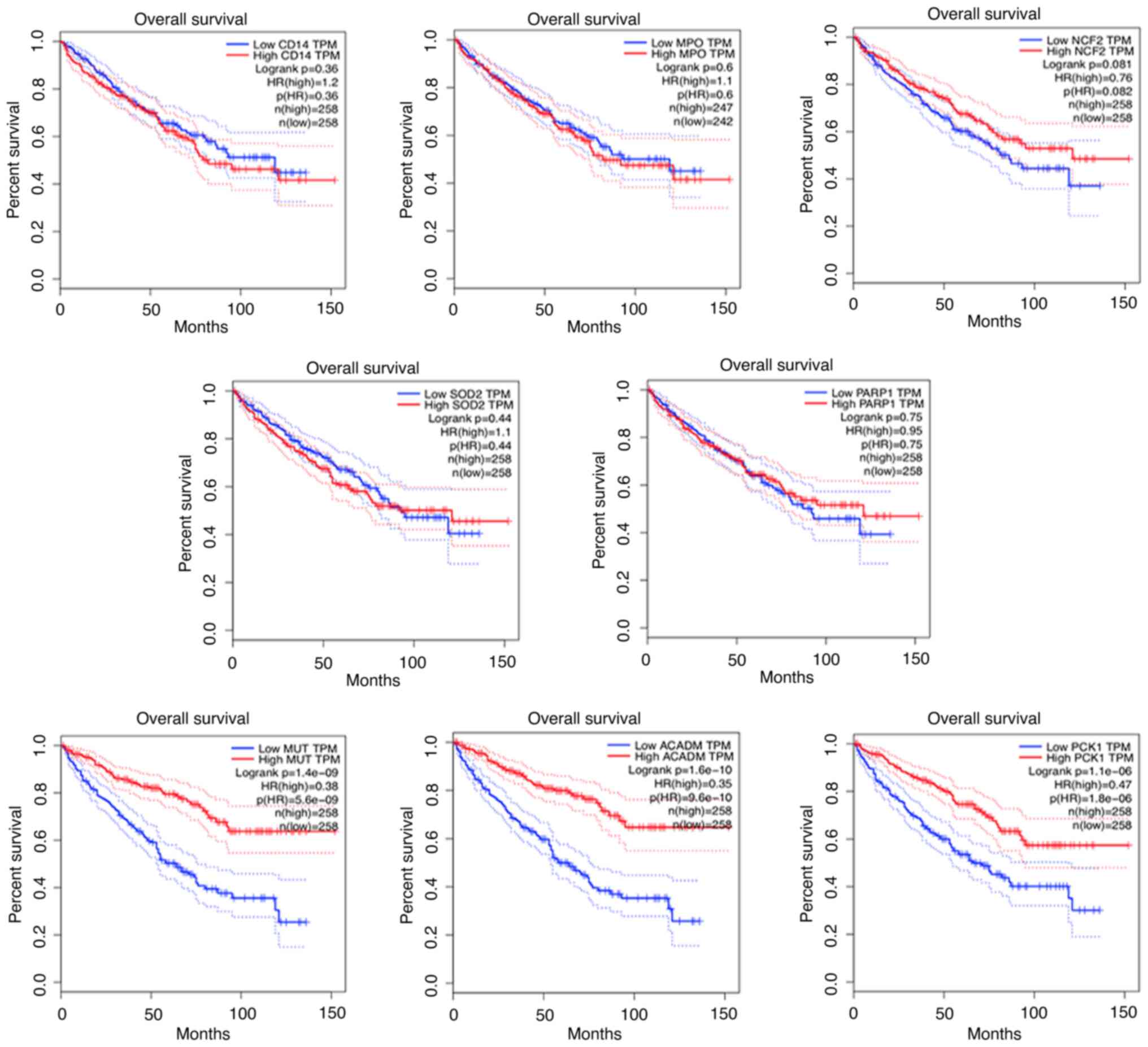
addition, we also analyzed the correlation between these proteins
and RCC prognosis by using online software GEPIA. Interestingly,
the upregulated proteins (CD14, MPO, NCF2, SOD2, PARP1) showed no
prognostic value in RCC patients, while prognostic value in the
downregulated proteins (MUT, ACADM, PCK1) were found (Fig. 10).
Discussion
In the present research, a quantitative proteomic
analysis was used to investigate the underlying molecular
mechanisms of renal cell carcinoma (RCC) development. We combined
both TMT labeling and LC-MS/MS-based enrichment to explore the
global proteins and post-translational modification (PTM) profiles
in tumor tissues. A total of 709 differentially expressed proteins
(DEPs) and 649 differentially expressed phosphorylated sites were
identified in the RCC tissues.
Our data underlined that the DEPs in the RCC tissues
were enriched in glycolysis, fatty acid metabolism and oxidative
phosphorylation. Changes in energy metabolism is known as a
biochemical feature of cancer cells, one of the hallmarks of
cancer. Many researchers have indicated that altered energy
metabolism is an important biological process of cancer cells
(18–20). Normal cells utilize glucose mainly in
the mitochondria to generate energy, while in cancer cells, energy
is largely generated through activated glycolysis in the cytosol in
hypoxic and acidic microenvironments. In addition, glycolysis still
remains the main approach of energy production for cancer cells
even in the presence of ample oxygen. Importantly, glycolysis plays
a crucial role in tumor cell metastasis, proliferation, and
resistance to chemotherapeutics (21–23).
In recent years, more and more researchers have
started to focus on the PTMs of proteins to explore the underlying
mechanisms in cancers (7,8). In addition to phosphorylation, many
other types of PTMs, for example, propionylation, acetylation,
succinylation, ubiquitylation and crotonylation have been
determined due to the development of mass spectrometry technology.
Importantly, the relationship between the PTMs in tumor progression
and regulating cellular energy metabolism have been demonstrated
(9).
Phosphorylation, an important type of PTM in
proteins, is widely distributed in diverse model organisms. Recent
research has revealed that global phosphorylation is dynamically
changed as a response to stress, changes in the microenvironment
and genetic mutations (12,24,25). At
the phosphorylation level, our results showed that numerous
metabolism-related processes were enriched in the RCC samples. The
upregulated phosphorylation proteins were mainly involved in
glucagon signaling pathway and cholesterol metabolism, while the
downregulated phosphorylation proteins were enriched in glycolysis,
pentose phosphate pathway, carbon metabolism and biosynthesis of
amino acids, showing a strong correlation between phosphorylation
and metabolic changes in RCC samples.
By using proteomic methods, we have offered a clear
insight into protein and phosphorylation profiles in human RCC.
These results can broaden our insight into RCC tumorigenesis and
progression, particularly the metabolism-related regulation of RCC.
In addition, our research identified various original potential
biomarkers for RCC. However, the effect of phosphorylation on
metabolism-related regulation and its specific regulated mechanisms
still needs to be rigorously verified by cell and animal
research.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant nos. 21402173, 81672520,
81773389 and 30973001); the Zhejiang Provincial Natural Science
Foundation of China (grant nos. LY17H160020 and LY17H050003); the
Zhejiang Province Medical Program (nos. 2014ZDA011 and 2014KYB134);
the National Basic Research 973 Program of China (no. 2012CB518304)
and the Health Department of Zhejiang Province (no. 2019ZD027).
Availability of data and materials
The authors of the present study have uploaded the
sequence data in public repositories (ProteomeXchange, project
accession, PXD025551).
Authors' contributions
LX and YC conceived and designed the study. LX ZL
and SY performed the experiments and wrote the paper. GL and YC
reviewed the data and edited the manuscript. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
These samples were obtained with the permission of
the Ethics Committee of Sir Run Run Shaw Hospital affiliated with
Zhejiang University Medical College (Hangzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Smith RA, Andrews K, Brooks D, DeSantis
CE, Fedewa SA, Lortet-Tieulent J, Manassaram-Baptiste D, Brawley OW
and Wender RC: Cancer screening in the United States, 2016: A
review of current American cancer society guidelines and current
issues in cancer screening. CA Cancer J Clin. 66:96–114. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Owens B: Kidney cancer. Nature.
537:S972016. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chow WH, Dong LM and Devesa SS:
Epidemiology and risk factors for kidney cancer. Nat Rev Urol.
7:245–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jonasch E, Gao J and Rathmell WK: Renal
cell carcinoma. BMJ. 349:g47972014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zarrabi K, Fang C and Wu S: New treatment
options for metastatic renal cell carcinoma with prior
anti-angiogenesis therapy. J Hematol Oncol. 10:382017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sánchez-Gastaldo A, Kempf E, González Del
Alba A and Duran I: Systemic treatment of renal cell cancer: A
comprehensive review. Cancer Treat Rev. 60:77–89. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Keskin O, Tuncbag N and Gursoy A:
Predicting protein-protein interactions from the molecular to the
proteome level. Chem Rev. 116:4884–4909. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Aebersold R and Mann M: Mass-spectrometric
exploration of proteome structure and function. Nature.
537:347–355. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Riley NM and Coon JJ: Phosphoproteomics in
the age of rapid and deep proteome profiling. Anal Chem. 88:74–94.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Silva AMN, Vitorino R, Domingues MRM,
Spickett CM and Domingues P: Post-translational modifications and
mass spectrometry detection. Free Radic Biol Med. 65:925–941. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Na S and Paek E: Software eyes for protein
post-translational modifications. Mass Spectrom Rev. 34:133–147.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Humphrey SJ, James DE and Mann M: Protein
phosphorylation: A major switch mechanism for metabolic regulation.
Trends Endocrinol Metab. 26:676–687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Butterfield DA, Gu L, Di Domenico F and
Robinson RA: Mass spectrometry and redox proteomics: Applications
in disease. Mass Spectrom Rev. 33:277–301. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Konstantinopoulos PA, Karamouzis MV and
Papavassiliou AG: Post-translational modifications and regulation
of the RAS superfamily of GTPases as anticancer targets. Nat Rev
Drug Discov. 6:541–555. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Okamura N, Masuda T, Gotoh A, Shirakawa T,
Terao S, Kaneko N, Suganuma K, Watanabe M, Matsubara T, Seto R, et
al: Quantitative proteomic analysis to discover potential
diagnostic markers and therapeutic targets in human renal cell
carcinoma. Proteomics. 8:3194–3203. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Masui O, White NM, DeSouza LV, Krakovska
O, Matta A, Metias S, Khalil B, Romaschin AD, Honey RJ, Stewart R,
et al: Quantitative proteomic analysis in metastatic renal cell
carcinoma reveals a unique set of proteins with potential
prognostic significance. Mol Cell Proteomics. 12:132–144. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Atrih A, Mudaliar MA, Zakikhani P, Lamont
DJ, Huang JT, Bray SE, Barton G, Fleming S and Nabi G: Quantitative
proteomics in resected renal cancer tissue for biomarker discovery
and profiling. Br J Cancer. 110:1622–1633. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gottlieb E and Mostoslavsky R: Cancer and
metabolism: Why should we care? Semin Cell Dev Biol. 43:1–2. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
DeBerardinis RJ and Chandel NS:
Fundamentals of cancer metabolism. Sci Adv. 2:e16002002016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cantor JR and Sabatini DM: Cancer cell
metabolism: One hallmark, many faces. Cancer Discov. 2:881–898.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wallace DC: Mitochondria and cancer. Nat
Rev Cancer. 12:685–698. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Andrejeva G and Rathmell JC: Similarities
and distinctions of cancer and immune metabolism in inflammation
and tumors. Cell Metab. 26:49–70. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ganapathy-Kanniappan S and Geschwind JF:
Tumor glycolysis as a target for cancer therapy: Progress and
prospects. Mol Cancer. 12:1522013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Swaney DL, Beltrao P, Starita L, Guo A,
Rush J, Fields S, Krogan NJ and Villén J: Global analysis of
phosphorylation and ubiquitylation cross-talk in protein
degradation. Nat Methods. 10:676–682. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Robles MS, Humphrey SJ and Mann M:
Phosphorylation is a central mechanism for circadian control of
metabolism and physiology. Cell Metab. 25:118–127. 2017. View Article : Google Scholar : PubMed/NCBI
|