Introduction
Hepatocellular carcinoma (HCC) is the most common
type of liver cancer, accounting for ~90% of all cases (1). HCC is one of the most common
malignancies and the third leading cause of cancer-related death
(2). Over 4 million estimated new
cases of HCC have been reported, and HCC has been reported to cause
over 3 million estimated deaths in USA (3). Multiple factors, including hepatitis B
and C, obesity, excess ingestion of alcohol and smoking, lead to
HCC progression. Apart from external factors, gene deficiency or
mutation is also recognized as a potential cause of liver cancer
progression (4–8). General treatments for HCC are surgery,
liver transplantation, and drug therapy. However, the overall
efficacy of HCC treatment remains unsatisfactory due to the high
recurrence and progression rates (9). Therefore, ongoing studies in HCC are
necessary. Complex molecular signaling pathways associated with
cell proliferation, metastasis, inflammatory reactions and drug
resistance are involved in HCC. The multiple types of molecules
involved in these pathways interact with each other and provide
various signal transduction pathways that are highly activated in
cancers (10–13).
HCC is the most common primary hepatic malignant
tumor and has been the 2nd most common cause of cancer-related
years of life lost worldwide. The prevalence in Taiwan ranks 4th
among all malignancies (~29/100,000) and it is the 2nd cause of
mortality (14). Most individuals
diagnosed with HCC often have chronic liver disease, particularly
chronic hepatitis B and C infection, and have not always symptoms
in the early stage. ~70% of patients cannot receive curative
therapy when they are diagnosed. The average survival time of
patients with HCC in Barcelona Clinic Liver Cancer (BCLC) stage C
is ~11 months, and that of patients in stage D is ~3 months.
In 2007, sorafenib was approved for HCC treatment.
The therapy of HCC entered a new era compared with traditional
chemotherapy, and sorafenib became a standard of care in the BCLC
stage C group. In the pivotal sorafenib HCC assessment randomized
protocol (SHARP) trial, sorafenib in patients with BCLC stage C HCC
showed an increased 2.8-month overall survival (OS) (10.7 months)
compared with placebo (7.9 months) (15). Before sorafenib, no significant
trial had shown a favorable response to treat patients with HCC by
traditional chemotherapy. Sorafenib is a multikinase inhibitor
(MKI) that suppresses tumor cell proliferation and angiogenesis
through different pathways (16).
However, in the last 10 years, other TKIs have failed to improve
the efficacy of sorafenib, including erlotinib, brivanib, sunitinib
and everolimus. Until 2017, regorafenib (REG) showed a significant
treatment response in patients with sorafenib-refractory disease
compared with placebo. In recent years, several TKIs have shown
treatment effects as 2nd-line (REG, cabozantinib, ramuciramab) and
1st-line (lenvatinib) agents, and the development of immune
checkpoint inhibitors (ICIs) has revealed another treatment option.
Systemic therapy for the BCLC stage C group or BCLC stage B group
refractory to transarterial embolization had more medication
choices in those years (17).
HCC patients with moderately compromised liver
function (Child-Pugh B functional status) have limited curative
options due to the risk of liver failure (18). This is particularly evident in HCC
cases not suitable for surgery or locoregional treatments. For
these patients, the availability of sorafenib differs, which is
differentiated by the different international guidelines and local
regulatory policies (19).
Previously, the potential mechanisms of metronomic capecitabine
(MC) have been mentioned, including blockage of tumor angiogenesis,
reduced therapeutic resistance, and activation of immune responses
(20–22). De Lorenzo et al (23) reported that the median OS of 35
MC-treated patients was 7.5 months [95% CI: 3.733-11.267] and 5.1
months [95% CI: 4.098-6.102] in the 70 BSC group (P=0.013)
(23). Furthermore, 12 patients
(34.3%) treated with MC experienced several adverse events,
including fatigue (17.1%), hand-foot syndrome (8.5%), neutropenia
(5.7%), and thrombocytopenia (8.5%). However, MC appears to be a
safe choice for Child-Pugh B-HCC patients. Its potential antitumor
activity warrants prospective evaluations (23). Similarly, MC also displayed a
therapeutic alternative for CP-B patients who were resistant to
tyrosine kinase inhibitors (TKIs).
Furthermore, radiation, chemotherapy, TKIs and ICIs
as adjuvant strategies have not been demonstrated to improve the
clinical status of progression-free survival (PFS) or OS in
resected early-stage disease (24).
Patients with early recurrence risk are usually associated with
negative prognostic factors, whereas those at risk of late
recurrence are always associated with advanced liver disease, such
as tumor formation (25). In fact,
some studies have shown that adjuvant systemic therapy can reduce
local and distant recurrence rates; however, more recent studies
have not demonstrated this effect (26).
Previously, Ohata et al (8) evaluated whether adjuvant systemic
treatment could improve survival in early-stage HCC, and the STORM
trial evaluated the effect of adjuvant sorafenib in HCC. In this
phase 3, double-blind study, 1,114 patients were randomly assigned
to placebo or sorafenib-stimulated groups, who were treated with
400 mg twice a day for up to 4 years. However, relapse-free
survival was not significantly different between the two groups
(27). Based on the evidence, it
was considered that the efficacy of anticancer agents cannot
translate from the advanced to the adjuvant setting. Unfortunately,
effective regimens thus far have not been found. At the same time,
because TKIs need an optional ‘target’ to perform their activity,
it was suggested that these drugs in the adjuvant setting are
probably far from being rational (10). Therefore, immunotherapy is
considered to be a promising approach, and patients should be
encouraged to enroll in clinical trials evaluating immune-based
combinations or ICI monotherapy as adjuvant treatment to reduce the
risk of recurrence and improve clinical outcomes.
REG, an orally bioavailable MKI, blocks the activity
of several protein kinases, including KIT, BRAF, RAF-1, RET,
VEGFR-1, VEGFR-2, VEGFR-3, PDGFR and FGFR, which are associated
with tumor microenvironment signaling, tumor angiogenesis, and cell
proliferation (28,29). Currently, REG is approved as a
single agent for the treatment of HCC at a dose of 160 mg orally
once daily on days 1–21 of each 28-day cycle (30). There are currently ongoing trials
aimed at evaluating the efficacy of REG as monotherapy or in
combination with other anticancer agents, and the number of
patients receiving REG is expected to increase in the future
(31,32). Therefore, it was suggested that REG
dose personalization may improve quality of life, decrease
treatment adverse events, and enhance patient outcomes.
ICIs are able to influence immune checkpoint-related
molecules, such as programmed cell death-1 (PD-1), cytotoxic
T-lymphocyte-associated antigen 4 (CTLA-4), and
lymphocyte-activation gene 3 (LAG-3) (33,34).
However, ICI monotherapy has demonstrated disappointing effects
thus far in patients with HCC (35,36).
Recently, combined immunotherapy with phase III IMbrave150 and the
PD-L1 inhibitor atezolizumab plus anti-angiogenesis bevacizumab
showed more convincing effects than monotherapy in advanced HCC
patients (37,38). Moreover, patients receiving another
combined immunotherapy, including atezolizumab and bevacizumab, had
impressive benefits in PFS, OS, objective response rate, and
complete response rate, and the median OS could reach 19.2 months
(39). However, to date, few
molecules have been assessed as predictors, such as programmed
death ligand-1 (PD-L1), tumor mutational burden, and gut
microbiota, to evaluate whether these therapeutic regimens benefit
patients with HCC (40). Hence, it
was suggested by the authors that a useful marker is needed to
evaluate whether monotherapy or combined immunotherapy benefits
patients with HCC.
The review entitled ‘The Roles of Protein Tyrosine
Phosphatases (PTP) in HCC’ published in 2018, reported both
oncogenic and tumor suppressive function of PTPs in HCC. Huang
et al (41) mainly reviewed
the involvement of PTP and associated signaling pathways in HCC.
However, the present review mainly focused on discussing the
relationship of PTPs with inflammatory cytokines and
chemotherapy/targeted drug resistance, providing detailed
information on how PTPs can modulate inflammatory reactions and
drug resistance to influence progression in HCC. The roles of
inflammatory cytokines and drug resistance modulated by PTPs have
never been displayed previously. Therefore, it was suggested that
the PTP-regulated inflammatory cytokines and drug resistance are
critical in HCC, and PTPs may be therapeutic targets in the future.
In the present review, numerous kinds of PTPs associated with
cancer progression were discussed, including those associated with
inflammatory reactions and drug resistance in HCC.
The PTP family
Class I PTPs include ~100 proteins, which can be
separated into two groups based on their interaction residues:
classical PTPs and dual-specificity phosphatases (DSPs). The
classical PTPs include ~38 proteins; these proteins are dispersed
in the cytoplasm (non-transmembrane PTPs) and cellular membrane
(receptor-like PTPs), and the classical PTPs are involved in a
broad range of pathways connecting intracellular and extracellular
components (42).
Tyrosine phosphate groups are the general site of
dephosphorylation of classical PTPs. By contrast, DSPs have three
phosphate-related areas, including tyrosine, serine and threonine
residues. In addition to DSPs having smaller catalytic domains than
classical PTPs, there are other major differences between classical
PTPs and DSPs. Most DSPs are localized in the cytoplasm, and due to
the diverse phosphate residues involved in the dephosphorylation
process, DSPs regulate various cellular biogenesis processes,
including the cell cycle, metabolism and neuron transduction
(43). Currently, DSPs are
categorized into several subclasses, such as mitogen-activated
protein kinase (MAPK) phosphatases (MPKs, 11 members), protein
phosphates slingshot homologs (3 members), phosphatases of
regenerating liver (PRLs, 3 members), phosphate and tension
homologs (PTEN, 5 members), atypical DSPs (19 members),
myotubularins (16 members), and CDC14s (4 members) (Table I) (43,44).
To date, certain of these DSP subclasses have been revealed to
potentially regulate pathways in multiple diseases, particularly
cancer. The MAPK/extracellular signal-regulated kinase (ERK)
pathway is one of the most important pathways, and its members have
been identified to be overexpressed or mutated in over 90% of
cancers. The target phosphatases of this pathway are MPKs.
Moreover, cancer cells are able to regulate CDK-induced cell cycle
progression through regulation of CDC14s; a previous study has
revealed upregulation of certain PRLs and downregulation in certain
PTEN members and myotubularins (45). Most class I PTPs induce profound
effects in cancers, while the characteristics of other genes and
proteins remain unclear (44).
Low-molecular-weight phosphatase, also called ACP1, is the only
member of the class II PTPs, and it has been demonstrated to be
associated with platelet-derived growth factor signaling (46).
 | Table I.The subclasses of DSPs. |
Table I.
The subclasses of DSPs.
| Subclass | Symbol | Member |
|---|
| Mitogen-activated
protein kinase phosphatases | MPKs | 11 |
| Phosphatases of
regenerating liver | PRLs | 3 |
| Phosphate and
tension homologs | PTEN | 5 |
| Atypical DSPs |
| 19 |
| Myotubularins |
| 16 |
| CDC14s |
| 4 |
PTP mechanisms
PTPs are multifunctional enzymes mainly localized in
the cytoplasm and cellular membrane. These enzymes play an
essential role in protein post-translational modification, which
affects protein activity and stability (47,48). A
common mechanism of PTPs is to recognize the phosphorylation
residues on tyrosine, serine, and threonine residues of target
proteins to induce the removal of phosphate groups, leading to the
dephosphorylation and inactivation of kinases or receptors
(49). Two PTP domains participate
in this process, the PTP-loop and WPD-loop. In the initiation of
the phosphorylation catalytic process, the cysteine catalytic site
on the PTP-loop competes for the binding of the phosphate group and
the target protein, which results in breaking of the
phosphate-oxygen bond and dephosphorylation of the target residue.
Concurrently, the WPD loop stabilizes the remaining structure. In
the further steps of the catalytic process, the phosphate group is
released via cooperation of the P-loop and WPD-loop (50).
Protein tyrosine phosphorylation is a pivotal
process for signal transduction in eukaryotic cells and is a
reversible regulatory mechanism that is coordinately controlled by
protein tyrosine kinases (PTKs) and PTPs (51). Imbalance of the PTK-PTP axis often
results in aberrant protein tyrosine phosphorylation in cancers and
promotes tumorigenesis, as is observed in HCC (52). PTPs usually play tumor-suppressor
roles, whereas PTKs are mainly associated with oncogenic and
tumorigenic activities (53).
Regulation of RTKs and PTPs, which occurs by reversible alteration
of the phosphorylation state of specific tyrosine kinases, leads to
various cellular events, such as alterations in the signaling
pathway activity and cellular phenotypes (54).
Association between PTPs and cytokines in
HCC
Cytokines have been identified as potential factors
in cancer progression and are components of the tumor
microenvironment. Cytokines include a broad range of extracellular
molecules, such as chemokines, interleukins, interferons and tumor
necrosis factors (TNFs). They are generally produced in the
extracellular environment by immune cells, endothelial cells and
stromal cells and regulate downstream cellular signaling by binding
to specific receptors (55). Due to
the enormous variety of cytokines and their varied characteristics,
the final cellular effects of cytokines are usually substantial and
long lasting. Cytokines can also increase cancer cell malignancy by
modifying cellular proliferation, migration and invasion (55). Several studies have demonstrated
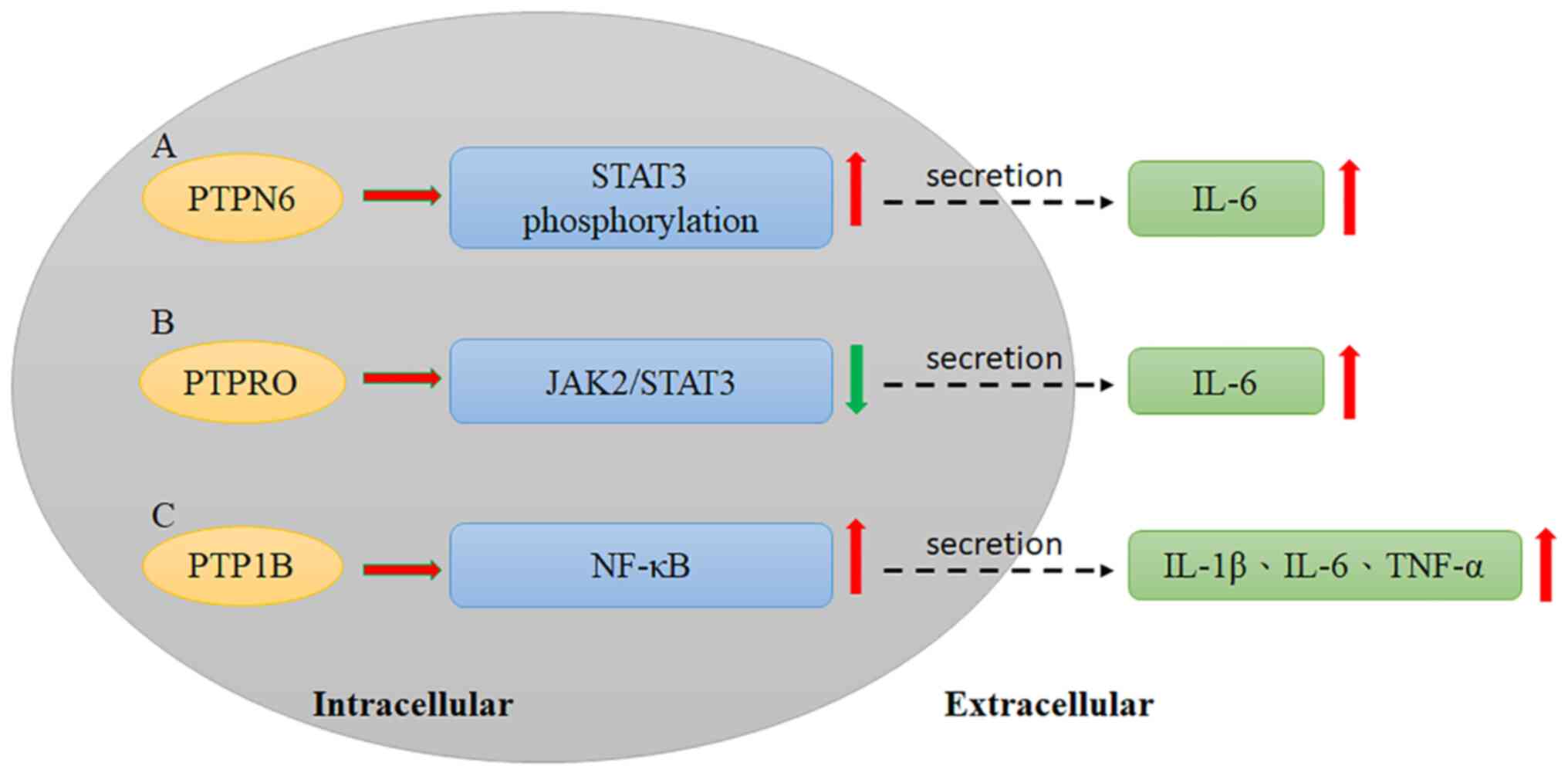
that some cytokines and PTPs interact in HCC progression. PTPN6
interacts with gankyrin, an oncoprotein, leading to the secretion
of IL-6, which induces signal transducer and activator of
transcription 3 (STAT3) phosphorylation and promotes HCC
development (56); Sakurai et
al (56) reported that gankyrin
can interact with PTPN6 to induce STAT3 activation and IL-6
secretion. Moreover, gankyrin can also increase VEGF expression,
which leads to HCC progression (Fig.
1A, Table II) (56). however, more studies have identified
PTPs as potential candidates for reversing cytokine-induced HCC
progression. PTP receptor type delta (PTPRD) is a tumor suppressor
that is negatively correlated with PD-L1, an essential molecule in
cancer immune escape. Overexpression of PTPRD is able to reduce
PD-L1 through the STAT3 pathway activation (Table II) (57). In addition, the effects of
combination therapies also involve PTP-cytokine relationships.
Induction of SHP-1 through crocin decreases the IL-6-stimulated
STAT3 pathway, resulting in HCC cell apoptosis (58). Quercetin enhances IFN-α-induced
phosphorylation of STAT1 by downregulating SHP2, leading to an
anti-proliferative effect in HCC (59).
| Table II.Association between PTPs and
cytokines. |
Table II.
Association between PTPs and
cytokines.
| Name | Mechanism | Pathway | Effect |
|---|
| PTPN6 | Interaction with
gankyrin ↑ | STAT3
phosphorylation ↑ | IL-6 secretion
↑ |
| PTPRD | Correlation with
PD-1 ↓ | STAT3 pathway
↑ | PD-L1 ↓ |
| CDC25A | IL-6 and IL-1β
↓ |
| Cell cycle
arrest |
| PTPRO |
| JAK2/STAT3 ↓ | IL-6 secretion ↑
PD-L1 ↓ |
| PTPN11 |
| RAS/ERK
pathway/Integrin signaling ↑ | HCC
progression |
| PTP1B | IL-1β, IL-6 and
TNF-α ↑ | NF-κB pathway
↑ | Inflammatory
response ↑ |
In addition to classical PTPs, DSPs are a subgroup
of tyrosine phosphatases in the PTP family, and these enzymes
function in the removal of a wide range of phosphate groups from
not only tyrosine residues but also serine/threonine residues
(44). Similar to classical PTPs,
DSPs are involved in broad signaling transduction in the regulation
of the development of both normal and cancer cells, and these
proteins also exhibit potential relationships with cytokines
(60). Sorafenib, the most common
targeted therapy in HCC treatment, induces DSP1 expression and
reduces TGF-β expression in macrophages, potentially promoting HCC
progression (61). Moreover,
knockdown of CDC25A decreases the expression of IL-6 and IL-1β,
leading to significant cell cycle arrest in the G1 phase in HCC
(62).
Previously, PTP receptor type O (PTPRO) and its
truncated form (PTPROt) were defined as negative regulators of
JAK2/STAT3 signaling (63). Hou
et al (63) reported that
PTPRO downregulates STAT3 activation via the JAK2 and PI3K
signaling pathways. Therefore, the effect of PTPRO on HCC
development may result from STAT3 activation. Numerous studies have
reported that the poor therapeutic effect of PD-1 adjuvant
treatment is highly associated with an increased level of IL-6 in
serum. Anti-PD-1/PD-L1 antibody combined with anti-IL-6 antibody
treatment has demonstrated significant curative effects in animal
models (64). Therefore, IL-6 may
have a crucial impact on the effect of PD-1/PD-L1 adjuvant therapy.
JAK2 has been demonstrated to interact with PTPRO upon IL-6
stimulation by a coimmunoprecipitation assay, indicating that PTPRO
may modulate JAK2 (Fig. 1B,
Table II). PD-L1 expression in
monocytes and macrophages has been demonstrated to be suppressed by
PTPRO through downregulation of the JAK2/STAT1 and JAK2/STAT3/c-MYC
cascades (65).
PD-L1 expression was significantly increased in
PTPRO-expressing macrophages in an IFN-γ-dependent manner after
IL-6 treatment for 72 h. This result could indicate that PTPRO
expression is essential for IL-6-induced Pd-L1/PD-L1 expression in
both Ptpro knockout and PTPRO knockdown macrophages.
Moreover, signaling pathway analysis indicated that the action of
PTPRO is dysregulated by IL-6 via increased activation of the
STAT3/c-MYC/PD-L1 axis in monocytes and macrophages. Treatment of
U937- and THP-1-derived macrophages with c-MYC shRNA reversed the
IL-6-induced decrease in PTPRO expression (65). IL-6 is secreted by both T cells and
macrophages and is a classic proinflammatory cytokine. IL-6 is a
key cytokine linking inflammation to tumorigenesis in numerous
cancers, including HCC (66,67).
Moreover, Naugler et al (68) also reported that IL-6 is an
essential cytokine linking inflammation and tumorigenesis. IL-6 has
been demonstrated to play an oncogenic role in obesity-related HCC
(69).
The nonreceptor PTP SHP2, encoded by PTP,
nonreceptor type 11 (PTPN11), is a critical member of the RAS/ERK
pathway and most receptor tyrosine kinase, cytokine receptor, and
integrin signaling pathways (70).
Several lines of evidence have indicated that PTPN11 is involved in
HCC progression (71). In addition,
several studies have shown that PTPN11 can also play an unexpected
tumor suppressor role in HCC (72,73),
implying that PTPN11 possesses dual roles in tumorigenesis.
PTPRD, a member of the PTP family, has been reported
to act as a tumor suppressor gene and plays a crucial role in
controlling numerous cellular processes, including cell
proliferation, apoptosis, survival and motility (74). PTPRD is often inactivated via
deletion or epigenetic mechanisms in several cancers (75). STAT3, a major transcription factor,
is involved in numerous cellular processes, such as cell growth,
proliferation, migration, differentiation and death (76). Previously, STAT3 has been
demonstrated to positively regulate PD-L1 expression to promote
immune escape in cancer (77).
Furthermore, Meng et al (57) reported that PTPRD expression was
significantly lower in tumor tissues than in normal tissues;
however, PD-L1 was significantly overexpressed in cancer tissues
compared with normal tissues.
Additional studies have also shown that silencing
PTP1B decreases the inflammatory response and levels of associated
cytokines, including IL-1β, IL-6 and TNF-α, while overexpression of
PTP1B induces inflammation in RAW264.7 cells. Moreover,
lipopolysaccharide can activate the NF-κB pathway in RAW264.7
cells, and NF-κB signaling is also affected by dysregulated PTP1B
expression (Fig. 1C, Table II) (78).
The nonreceptor PTP Src homology region 2 (SH2)
domain-containing phosphatases (SHPs), including SHP-1 (also known
as PTPN6) and SHP-2 (also known as PTPN11), are critical modulators
of numerous fundamental cellular processes, such as cell
proliferation, differentiation and inflammation (79). SHP-2 is a ubiquitously expressed
modulator of inflammatory reactions and is implicated in HCC
carcinogenesis and progression (80). In addition, SHP-1 is extensively
expressed in hematopoietic and epithelial cells and is widely
defined as a negative regulator of inflammation (81). Several studies have reported that
MKIs, such as sorafenib (82) and
dovitinib (83), exert their
antitumor effects by enhancing SHP-1 phosphatase activity.
TGF-β1-induced STAT3 (Tyr705) phosphorylation and
epithelial-to-mesenchymal transition can be abolished with SHP-1
overexpression, which blocks cell migration and invasion of HCC
(84). Moreover, SHP-1 has been
demonstrated to be overexpressed in non-cancer tissues compared
with surrounding cancer tissues, and reduced SHP-1 expression is
highly associated with poor prognosis of patients with HCC
(85). Collectively, SHP-1 can be
defined as a tumor suppressor that prevents the initiation and
progression of HCC in animal models (85). SHP-1 can also inhibit the activation
of various signaling pathways, such as the STAT3, NF-kB, and AKT
pathways, to suppress hepatocarcinogenesis and the malignant
phenotype of HCC (85). Several
drugs, including sorafenib, dovitinib, and SC-2001, induce cell
apoptosis and inhibit the growth of HCC cells by enhancing the
activity of SHP-1 tyrosine phosphatase (82,86).
SHP-1 and SHP-2, cytoplasmic PTPs, share similar sequences,
containing two Src homology 2 (SH2) NH2-terminal domains and a
C-terminal protein-tyrosine phosphatase domain (87). SHP-2 reduces STAT3 phosphorylation
via the JAK/STAT pathway to suppress HCC initiation. By contrast,
SHP-2 coordinately activates the Ras/Raf/Erk and PI3K/Akt/mTOR
cascades to promote the progression of HCC (80). Liver inflammation, a primary
oncogenic factor, is highly associated with HCC (88,89).
The inflammatory cytokines induced by liver injury activate
inflammatory signaling pathways, including the JAK/STAT and NF-kB
signaling pathways, which in turn induce the expression of IL6,
TGF-β, and TNF-α (90,91).
The relationship between PTPs and drug
resistance
SHP2 is related to the stress sensor DNA damage 45G
(GADD45G), which is involved in multiple biological processes and
downregulated in various cancers. GADD45G has been demonstrated to
induce senescence in HCC and reduce tumor growth in vivo.
Moreover, GADD45G-induced senescence can be efficiently
counteracted with Shp2 silencing. GADD45G expression is negatively
correlated with the phosphorylation status of STAT3 in tumor cells
of clinical HCC specimens (Table
III) (92); this result is
related to the relationship of SHP2 with STAT3 (93,94).
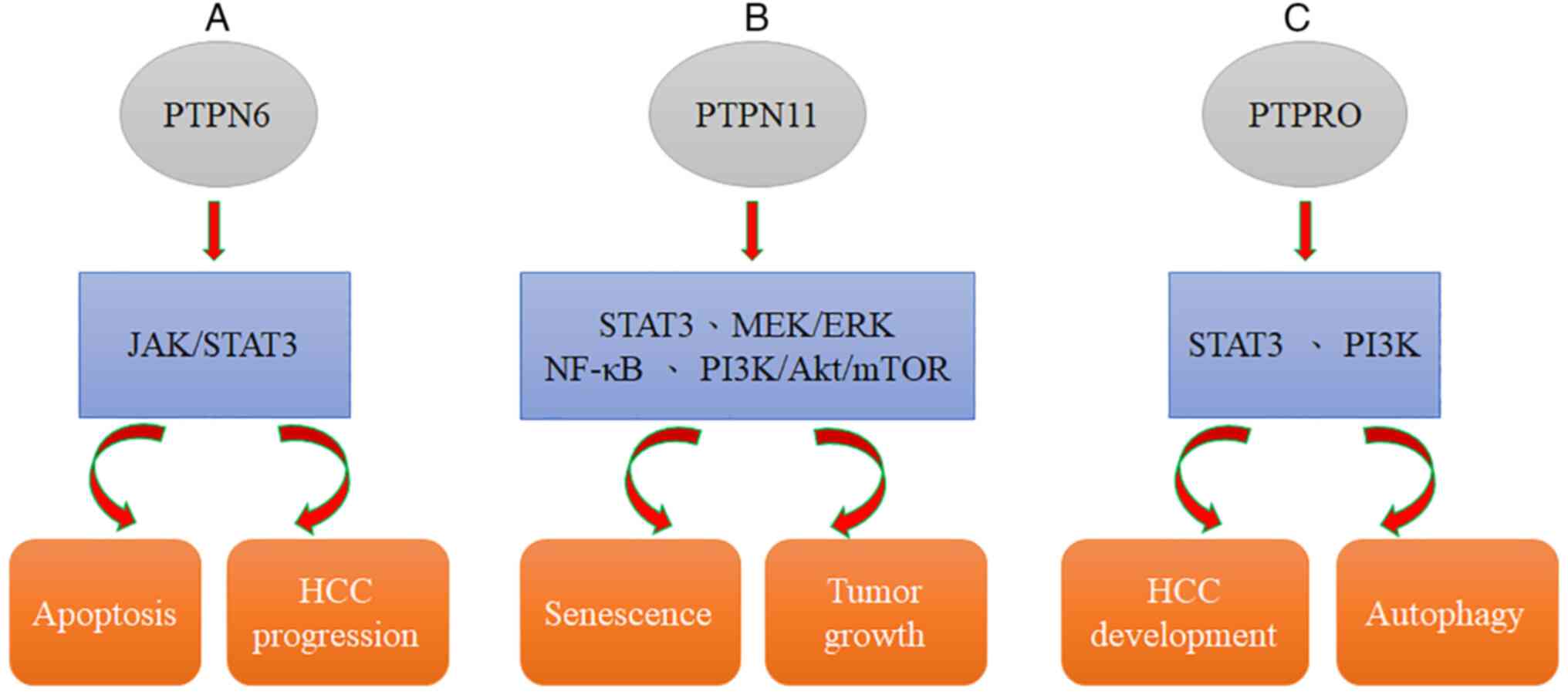
Dovitinib downregulation of p-STAT3 and induction of apoptosis can
be abolished by using an SHP-1 inhibitor or silencing SHP-1 with
RNA interference, suggesting that SHP-1, a PTP, modulates the
effects of dovitinib. In addition, dovitinib reduced STAT3
activation to induce cell apoptosis in two sorafenib-resistant cell
lines, and sorafenib-resistant cells showed significant activation
of STAT3, indicating that STAT3 may be a useful target to overcome
drug resistance in HCC (54).
JAK/STAT3 signaling is inactivated by several PTPs, including the
SH2 domain-containing cytosolic phosphatases SHP-1 and SHP-2
(95,96). Furthermore, SHP-1 has been
demonstrated to be involved in the dovitinib-mediated effect on
kinase inhibition, phosphorylated (p)-STAT3 and apoptosis in HCC
(54). Additionally, dovitinib
suppressed tumor growth in both Huh-7 and PLC5 ×enograft tumors
in vivo, suggesting the potential utility of dovitinib in
the clinical practice. Therefore, an understanding of the mechanism
of SHP-1-mediated STAT3 inhibition provides a potential target for
future HCC molecular therapy (Fig.
2A, Table III) (54).
| Table III.The relationship between PTPs and
drug resistance. |
Table III.
The relationship between PTPs and
drug resistance.
| PTPs | Markers | Pathway | Effect |
|---|
| PTPN6 | p-JAK1, p-JAK2,
Mcl-1, and cyclin D1 | JAK/STAT3 pathway
↑ | Apoptosis ↑ HCC
progression ↑ |
| PTPN11 | GADD45G | STAT3 pathway
↑ | Senescence ↑ |
|
|
| MEK/ERK pathways
↑ | Tumor growth ↓ |
|
|
| NF-κB pathway
↑ |
|
|
|
| PI3K/AKT/mTOR
pathway ↑ |
|
| PTPRO |
| STAT3 pathway
↓ | HCC development
↓ |
|
| LC3II/I, p62 | PI3K pathway ↓ | Autophagy ↓ |
SHP2, encoded by PTPN11, was found to not only be
overexpressed in HCC (97) but also
serve as a potential predictive biomarker for sorafenib response
and patient survival (97).
Moreover, SHP2 has been defined as a downstream effector of
numerous RTKs, and SHP2 blockade may be a possible mechanism
causing RTK activation, resulting in the development of acquired
resistance to sorafenib in HCC (98). Collectively, sorafenib-induced
reactivation of the RTK-mediated AKT and MEK/ERK pathways can be
significantly induced by SHP099 (98). Targeting SHP2 with SHP099 combined
with sorafenib treatment may be a novel and safe therapeutic
strategy against HCC (99).
Previously, Kang et al (100) reported that the RNA level of SHP2
is upregulated through the NF-κB signaling pathway in
HBX-transfected HCC cells. Mechanistically, SHP2 expression is
induced by direct binding of NF-κB to its promoter. Since NF-κB
signaling has been implicated in HCC progression (100) and sorafenib resistance (101), its activation may be a potent
mechanism leading to SHP2 upregulation in both parental and
sorafenib-resistant HCC cells. Previously, SHP2 was identified as
an oncogenic tyrosine phosphatase that contains two Src-homology 2
domains (102). SHP2 has been
reported to be a critical component of multiple RTK signaling
pathways activated in response to numerous growth factors,
including FGFR, EGFR, PDGFR, and VEGFR, and this activation leads
to induction of the PI3K/AKT/mTOR pathway and ERK signaling based
on genetic and biochemical evidence (103,104). SHP2 is required for RTK-evoked RAS
activation, which results in the activation of the MEK/ERK and AKT
pathways (Fig. 2B, Table III) (105).
SHP-1 is a PTP that is largely expressed in
hematopoietic cells. To date, several studies have addressed the
role of SHP-1 in tumor progression, and a few studies have
suggested that SHP1 plays a potential tumor suppressor role in
various cancer types (106).
Moreover, impaired function of SHP-1 has been shown to induce
cancer progression by downregulating intracellular signaling
transmembrane receptors, such as growth factor and cytokine
receptors, leading to abnormal pathologies (107). Upregulation of SHP-1 activity
induces cell apoptosis both in vitro and in vivo
(54). In addition, Tai et
al (54) reported that
STAT3-related kinases or downstream effectors, including p-JAK1,
p-JAK2, Mcl-1, and cyclin D1, are also induced in
sorafenib-resistant cells. Evidence has shown that the JAK/STAT3
signaling pathway is a crucial modulator of the efficacy of
sorafenib. Notably, decreased expression of SHP-1 was also observed
in sorafenib-resistant cells. Collectively, the role of
SHP-1-related STAT3 signaling in HCC has been verified; therefore,
the SHP-1/STAT3 pathway may be an effective target for HCC
treatment (54). SHP-1-mediated
dephosphorylation of PKM2 at Y105 results in increased activity,
and tetrameric PKM2 has reduced nuclear localization, which leads
to the downregulation of the expression of oncogenic molecules,
such as c-Myc and cyclin D1. Furthermore, constitutively active
SHP-1 (D61A) can increase the percentage of tetrameric PKM2 and
phosphorylation of PKM2 (Y105F), suggesting that SHP-1 determines
the levels of dimeric/tetramer PKM2 and the subsequent nuclear
localization via PKM2 Y105 dephosphorylation (108). SHP-1 (PTPN6), first
identified in hematopoietic cells, is implicated in various
hematopoietic signaling processes, including integration of
immunoreceptor tyrosine-based activation motif-mediated inhibitory
signals (109) and B-cell and
natural killer (NK)-cell development (110,111). Furthermore, gankyrin is
upregulated in chronic inflammation and induces STAT3 activation
and IL-6 secretion by interacting with SHP-1 in non-parenchymal
cells. Such proinflammatory interactions may induce the levels of
stem cell markers in the tumor microenvironment and eventually
promote HCC progression. Thus, the expression of gankyrin is
defined as a promising predictor of the efficacy of advanced
treatment for patients with HCC (56). Previously, gankyrin was demonstrated
to enhance hepatocarcinogenesis via STAT3 activation through SHP-1
inhibition and IL-6 upregulation in the tumor microenvironment.
Thus, STAT3/IL-6 signaling may involve gankyrin-regulated crosstalk
between tumor cells and nonparenchymal cells (56).
PTPRO is a receptor type of PTP that has been
defined as an integral membrane protein in numerous parenchymal
cells (including lung, liver, and breast cells) (112,113). It has been previously demonstrated
by the authors that PTPRO can suppress STAT3 activation, leading to
reduced development of HCC. PTPRO can negatively regulate important
pathways related to autophagy, such as the PI3K signaling pathway
(63,114). Moreover, Zhang et al
(115) reported that
ptpro−/− hepatocytes lead to the development of
steatosis and induce tumorigenesis in mice fed a high-fat diet
(HFD). PTPRO deletion significantly augmented obesity-reduced
autophagy, as evidenced by increased p62 expression and a reduced
LC3II/I ratio, as revealed by western blotting. Collectively,
evidence confirmed that PTPRO deletion promotes obesity-related
hyperinsulinemia and autophagy deficiency in the liver (Fig. 2C, Table III) (115). Furthermore, evidence suggested
that PTPRO increases the cytoplasmic accumulation of p53 through
the PI3K/Akt/MDM axis (115).
Additionally, the expression of PTPRO has been demonstrated to be
significantly reduced in HCC compared with normal tissues (63). PTPRO expression is suppressed in
vivo in mice fed an HFD compared with that in mice fed a normal
diet (116,117). PTPRO regulates autophagy and lipid
metabolism in obesity and steatohepatitis (115). Furthermore, PTPRO regulates lipid
metabolism through reduced expression of lipogenesis genes and
induction of β-oxidation-related gene expression, and obesity
significantly induces tumorigenesis in the liver in
ptpro−/− mice (115).
In the present review, extensive information was
provided discussing whether PTPs play a critical role in
inflammatory reactions and drug resistance to influence cancer
progression in HCC. Numerous inflammatory cytokines/chemokines
modulated by PTPs and several chemotherapeutic and targeted
therapeutic drugs were illustrated, that are likely related to PTPs
that play critical roles in numerous cellular mechanisms and
signaling pathways. A total of 3 PTPs involved in HCC drug
resistance were listed, including PTPN6, PTPN11 and PTPRO. In
addition, there are six PTPs, PTPN6, PTPRD, CDC25A, PTPRO, PTPN11
and PTP1B. Among these, it was found that the three PTPs PTPN6,
PTPN11 and PTPRO both induce drug resistance and alter inflammatory
cytokine regulation, and these molecules can influence tumor growth
and HCC progression. Hence, it was suggested that the three PTPs
PTPN6, PTPN11 and PTPRO play equally important roles in HCC
progression. The present review provided practical information for
researchers to understand in an improved way the roles and
functions of PTPs in cancer progression and hence may aid the
identification of further therapeutic options to cure cancer.
According to findings from other groups about the
roles of PTPs, it was suggested by the authors that PTPs play roles
in inflammatory cytokines and drug resistance. In the future, it is
considered by the authors that the effects of PTPs may be applied
to clinical practice to evaluate whether PTP molecules could be
useful predictors in patients with HCC. It was hypothesized that
the levels of PTPs and inflammatory cytokines, such as IL-1β and
IL-6, in HCC patients with chemotherapy/targeted drug resistance
could be detected in tissues and plasma by immunohistochemistry and
ELISA, respectively. Finally, the correlations between the levels
of PTPs and IL-1β and IL-6 can also be analyzed to determine
whether the correlations and these molecules could be markers to
predict the prognosis and survival rate of drug-resistant
patients.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Ministry of Science and
Technology of the Republic of China (grant nos. MOST
109-2320-B-006-067. and MOST 110-2320-B-039-044).
Availability of data and materials
Not applicable.
Authors' contributions
Y-LC, J-YC, Y-CH and C-YC performed formal analysis.
Y-LC, C-CH, J-YC and C-YC prepared the original draft of the
manuscript. C-CH, P-MC, Y-CH and C-Y wrote, reviewed and edited the
manuscript. P-MC and C-YC acquired funding. All authors have read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ringelhan M, Pfister D, O'Connor T,
Pikarsky E and Heikenwalder M: The immunology of hepatocellular
carcinoma. Nat Immunol. 19:222–232. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang JD and Roberts LR: Hepatocellular
carcinoma: A global view. Nat Rev Gastroenterol Hepatol. 7:448–458.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bosch FX, Ribes J, Diaz M and Cleries R:
Primary liver cancer: Worldwide incidence and trends.
Gastroenterology. 127 (5 Suppl 1):S5–S16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Morgan TR, Mandayam S and Jamal MM:
Alcohol and hepatocellular carcinoma. Gastroenterology. 127 (5
Suppl 1):S87–S96. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Caldwell SH, Crespo DM, Kang HS and
Al-Osaimi AM: Obesity and hepatocellular carcinoma.
Gastroenterology. 127 (5 Suppl 1):S97–S103. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ming L, Thorgeirsson SS, Gail MH, Lu P,
Harris CC, Wang N, Shao Y, Wu Z, Liu G, Wang X and Sun Z: Dominant
role of hepatitis B virus and cofactor role of aflatoxin in
hepatocarcinogenesis in Qidong, China. Hepatology. 36:1214–1220.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu MC and Yuan JM: Environmental factors
and risk for hepatocellular carcinoma. Gastroenterology. 127 (5
Suppl 1):S72–S78. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ohata K, Hamasaki K, Toriyama K, Matsumoto
K, Saeki A, Yanagi K, Abiru S, Nakagawa Y, Shigeno M, Miyazoe S, et
al: Hepatic steatosis is a risk factor for hepatocellular carcinoma
in patients with chronic hepatitis C virus infection. Cancer.
97:3036–3043. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Spangenberg HC, Thimme R and Blum HE:
Targeted therapy for hepatocellular carcinoma. Nat Rev
Gastroenterol Hepatol. 6:423–432. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Suriawinata A and Xu R: An update on the
molecular genetics of hepatocellular carcinoma. Semin Liver Dis.
24:77–88. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Satyanarayana A, Manns MP and Rudolph KL:
Telomeres and telomerase: A dual role in hepatocarcinogenesis.
Hepatology. 40:276–283. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brechot C: Pathogenesis of hepatitis B
virus-related hepatocellular carcinoma: Old and new paradigms.
Gastroenterology. 127 (5 Suppl 1):S56–S61. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ahn JH, Kim SJ, Park WS, Cho SY, Ha JD,
Kim SS, Kang SK, Jeong DG, Jung SK, Lee SH, et al: Synthesis and
biological evaluation of rhodanine derivatives as PRL-3 inhibitors.
Bioorg Med Chem Lett. 16:2996–2999. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: Sorafenib in advanced hepatocellular carcinoma. N Engl J
Med. 359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu YJ, Zheng B, Wang HY and Chen L: New
knowledge of the mechanisms of sorafenib resistance in liver
cancer. Acta Pharmacol Sin. 38:614–622. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Villanueva A: Hepatocellular Carcinoma. N
Engl J Med. 380:1450–1462. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Granito A and Bolondi L: Non-transplant
therapies for patients with hepatocellular carcinoma and
Child-Pugh-Turcotte class B cirrhosis. Lancet Oncol. 18:e101–e112.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tovoli F, Negrini G and Bolondi L:
Comparative analysis of current guidelines for the treatment of
hepatocellular carcinoma. Hepat Oncol. 3:119–136. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tovoli F, Lorenzo S, Barbera MA, Garajova
I, Frega G, Palloni A, Pantaleo MA, Biasco G and Brandi G:
Postsorafenib systemic treatments for hepatocellular carcinoma:
Questions and opportunities after the regorafenib trial. Future
Oncol. 13:1893–1905. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pasquier E, Kavallaris M and Andre N:
Metronomic chemotherapy: New rationale for new directions. Nat Rev
Clin Oncol. 7:455–465. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kareva I, Waxman DJ and Lakka Klement G:
Metronomic chemotherapy: An attractive alternative to maximum
tolerated dose therapy that can activate anti-tumor immunity and
minimize therapeutic resistance. Cancer Lett. 358:100–106. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
De Lorenzo S, Tovoli F, Barbera MA, Garuti
F, Palloni A, Frega G, Garajovà I, Rizzo A, Trevisani F and Brandi
G: Metronomic capecitabine vs. best supportive care in Child-Pugh B
hepatocellular carcinoma: A proof of concept. Sci Rep. 8:99972018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Personeni N and Rimassa L: Hepatocellular
carcinoma: A global disease in need of individualized treatment
strategies. J Oncol Pract. 13:368–369. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Akateh C, Black SM, Conteh L, Miller ED,
Noonan A, Elliott E, Pawlik TM, Tsung A and Cloyd JM: Neoadjuvant
and adjuvant treatment strategies for hepatocellular carcinoma.
World J Gastroenterol. 25:3704–3721. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rizzo A, Mollica V, Ricci AD, Maggio I,
Massucci M, Rojas Limpe FL, Fabio FD and Ardizzoni A: Third- and
later-line treatment in advanced or metastatic gastric cancer: A
systematic review and meta-analysis. Future Oncol. 16:4409–4418.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bruix J, Takayama T, Mazzaferro V, Chau
GY, Yang J, Kudo M, Cai J, Poon RT, Han KH, Tak WY, et al: Adjuvant
sorafenib for hepatocellular carcinoma after resection or ablation
(STORM): A phase 3, randomised, double-blind, placebo-controlled
trial. Lancet Oncol. 16:1344–1354. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wilhelm SM, Dumas J, Adnane L, Lynch M,
Carter CA, Schütz G, Thierauch KH and Zopf D: Regorafenib (BAY
73-4506): A new oral multikinase inhibitor of angiogenic, stromal
and oncogenic receptor tyrosine kinases with potent preclinical
antitumor activity. Int J Cancer. 129:245–255. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Abou-Elkacem L, Arns S, Brix G, Gremse F,
Zopf D, Kiessling F and Lederle W: Regorafenib inhibits growth,
angiogenesis, and metastasis in a highly aggressive, orthotopic
colon cancer model. Mol Cancer Ther. 12:1322–1331. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rimassa L, Pressiani T, Personeni N and
Santoro A: Regorafenib for the treatment of unresectable
hepatocellular carcinoma. Expert Rev Anticancer Ther. 17:567–576.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cerrito L, Ponziani FR, Garcovich M,
Tortora A, Annicchiarico BE, Pompili M, Siciliano M and Gasbarrini
A: Regorafenib: A promising treatment for hepatocellular carcinoma.
Expert Opin Pharmacother. 19:1941–1948. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu Z, Lin Y, Zhang J, Zhang Y, Li Y, Liu
Z, Li Q, Luo M, Liang R and Ye J: Molecular targeted and immune
checkpoint therapy for advanced hepatocellular carcinoma. J Exp
Clin Cancer Res. 38:4472019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ingles Garces AH, Au L, Mason R, Thomas J
and Larkin J: Building on the anti-PD1/PD-L1 backbone: Combination
immunotherapy for cancer. Expert Opin Investig Drugs. 28:695–708.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Alsaab HO, Sau S, Alzhrani R, Tatiparti K,
Bhise K, Kashaw SK and Iyer AK: PD-1 and PD-L1 checkpoint signaling
inhibition for cancer immunotherapy: Mechanism, combinations, and
clinical outcome. Front Pharmacol. 8:5612017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cheng AL, Hsu C, Chan SL, Choo SP and Kudo
M: Challenges of combination therapy with immune checkpoint
inhibitors for hepatocellular carcinoma. J Hepatol. 72:307–319.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pinter M, Jain RK and Duda DG: The current
landscape of immune checkpoint blockade in hepatocellular
carcinoma: A review. JAMA Oncol. 7:113–123. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux
M, Kim TY, Kudo M, Breder V, Merle P, Kaseb AO, et al: Atezolizumab
plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J
Med. 382:1894–1905. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kelley RK: Atezolizumab plus Bevacizumab-A
landmark in liver cancer. N Engl J Med. 382:1953–1955. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rizzo A, Ricci AD and Brandi G:
Atezolizumab in advanced hepatocellular carcinoma: Good things come
to those who wait. Immunotherapy. 13:637–644. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sangro B, Sarobe P, Hervas-Stubbs S and
Melero I: Advances in immunotherapy for hepatocellular carcinoma.
Nat Rev Gastroenterol Hepatol. 18:525–543. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huang Y, Zhang Y, Ge L, Lin Y and Kwok HF:
The roles of protein tyrosine phosphatases in hepatocellular
carcinoma. Cancers (Basel). 10:822018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hendriks WJ, Elson A, Harroch S, Pulido R,
Stoker A and den Hertog J: Protein tyrosine phosphatases in health
and disease. FEBS J. 280:708–730. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pulido R and Hooft van Huijsduijnen R:
Protein tyrosine phosphatases: Dual-specificity phosphatases in
health and disease. FEBS J. 275:848–866. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Alonso A, Sasin J, Bottini N, Friedberg I,
Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J and Mustelin
T: Protein tyrosine phosphatases in the human genome. Cell.
117:699–711. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Buj-Bello A, Laugel V, Messaddeq N,
Zahreddine H, Laporte J, Pellissier JF and Mandel JL: The lipid
phosphatase myotubularin is essential for skeletal muscle
maintenance but not for myogenesis in mice. Proc Natl Acad Sci USA.
99:15060–15065. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chiarugi P, Cirri P, Marra F, Raugei G,
Fiaschi T, Camici G, Manao G, Romanelli RG and Ramponi G: The Src
and signal transducers and activators of transcription pathways as
specific targets for low molecular weight phosphotyrosine-protein
phosphatase in platelet-derived growth factor signaling. J Biol
Chem. 273:6776–6785. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hunter T and Sefton BM: Transforming gene
product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl
Acad Sci USA. 77:1311–1315. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cohen P: Protein kinases-the major drug
targets of the twenty-first century? Nat Rev Drug Discov.
1:309–315. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tanner JJ, Parsons ZD, Cummings AH, Zhou H
and Gates KS: Redox regulation of protein tyrosine phosphatases:
Structural and chemical aspects. Antioxid Redox Signal. 15:77–97.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang ZY: Protein tyrosine phosphatases:
Structure and function, substrate specificity, and inhibitor
development. Annu Rev Pharmacol Toxicol. 42:209–234. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
He RJ, Yu ZH, Zhang RY and Zhang ZY:
Protein tyrosine phosphatases as potential therapeutic targets.
Acta Pharmacol Sin. 35:1227–1246. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang ZC, Gao Q, Shi JY, Guo WJ, Yang LX,
Liu XY, Liu LZ, Ma LJ, Duan M, Zhao YJ, et al: Protein tyrosine
phosphatase receptor S acts as a metastatic suppressor in
hepatocellular carcinoma by control of epithermal growth factor
receptor-induced epithelial-mesenchymal transition. Hepatology.
62:1201–1214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Meeusen B and Janssens V: Tumor
suppressive protein phosphatases in human cancer: Emerging targets
for therapeutic intervention and tumor stratification. Int J
Biochem Cell Biol. 96:98–134. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tai WT, Cheng AL, Shiau CW, Liu CY, Ko CH,
Lin MW, Chen PJ and Chen KF: Dovitinib induces apoptosis and
overcomes sorafenib resistance in hepatocellular carcinoma through
SHP-1-mediated inhibition of STAT3. Mol Cancer Ther. 11:452–463.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang CI, Chu PM, Chen YL, Lin YH and Chen
CY: Chemotherapeutic drug-regulated cytokines might influence
therapeutic efficacy in HCC. Int J Mol Sci. 22:136272021.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sakurai T, Yada N, Hagiwara S, Arizumi T,
Minaga K, Kamata K, Takenaka M, Minami Y, Watanabe T, Nishida N and
Kudo M: Gankyrin induces STAT3 activation in tumor microenvironment
and sorafenib resistance in hepatocellular carcinoma. Cancer Sci.
108:1996–2003. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Meng Q, Tian J, Qin F, Huang X, Zhu D,
Xiang B and Dong D: Protein tyrosine phosphatase receptor type
delta (PTPRD) suppresses the expression of PD-L1 in human
hepatocellular carcinoma by down-regulating STAT3. Transl Cancer
Res. 9:5574–5584. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kim B and Park B: Saffron carotenoids
inhibit STAT3 activation and promote apoptotic progression in
IL-6-stimulated liver cancer cells. Oncol Rep. 39:1883–1891.
2018.PubMed/NCBI
|
|
59
|
Igbe I, Shen XF, Jiao W, Qiang Z, Deng T,
Li S, Liu WL, Liu HW, Zhang GL and Wang F: Dietary quercetin
potentiates the antiproliferative effect of interferon-α in
hepatocellular carcinoma cells through activation of JAK/STAT
pathway signaling by inhibition of SHP2 phosphatase. Oncotarget.
8:113734–113748. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hammer M, Mages J, Dietrich H, Servatius
A, Howells N, Cato AC and Lang R: Dual specificity phosphatase 1
(DUSP1) regulates a subset of LPS-induced genes and protects mice
from lethal endotoxin shock. J Exp Med. 203:15–20. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wei X, Tang C, Lu X, Liu R, Zhou M, He D,
Zheng D, Sun C and Wu Z: MiR-101 targets DUSP1 to regulate the
TGF-β secretion in sorafenib inhibits macrophage-induced growth of
hepatocarcinoma. Oncotarget. 6:18389–18405. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chen S, Tang Y, Yang C, Li K, Huang X and
Cao J: Silencing CDC25A inhibits the proliferation of liver cancer
cells by downregulating IL6 in vitro and in vivo. Int J Mol Med.
45:743–752. 2020.PubMed/NCBI
|
|
63
|
Hou J, Xu J, Jiang R, Wang Y, Chen C, Deng
L, Huang X, Wang X and Sun B: Estrogen-sensitive PTPRO expression
represses hepatocellular carcinoma progression by control of STAT3.
Hepatology. 57:678–688. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tsukamoto H, Fujieda K, Miyashita A,
Fukushima S, Ikeda T, Kubo Y, Senju S, Ihn H, Nishimura Y and
Oshiumi H: Combined Blockade of IL6 and PD-1/PD-L1 signaling
abrogates mutual regulation of their immunosuppressive effects in
the tumor microenvironment. Cancer Res. 78:5011–5022. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhang W, Liu Y, Yan Z, Yang H, Sun W, Yao
Y, Chen Y and Jiang R: IL-6 promotes PD-L1 expression in monocytes
and macrophages by decreasing protein tyrosine phosphatase receptor
type O expression in human hepatocellular carcinoma. J Immunother
Cancer. 8:e0002852020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
He G and Karin M: NF-κB and STAT3-key
players in liver inflammation and cancer. Cell Res. 21:159–168.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang K and Karin M: Tumor-Elicited
inflammation and colorectal cancer. Adv Cancer Res. 128:173–196.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Naugler WE, Sakurai T, Kim S, Maeda S, Kim
K, Elsharkawy AM and Karin M: Gender disparity in liver cancer due
to sex differences in MyD88-dependent IL-6 production. Science.
317:121–124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Park EJ, Lee JH, Yu GY, He G, Ali SR,
Holzer RG, Osterreicher CH, Takahashi H and Karin M: Dietary and
genetic obesity promote liver inflammation and tumorigenesis by
enhancing IL-6 and TNF expression. Cell. 140:197–208. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Grossmann KS, Rosario M, Birchmeier C and
Birchmeier W: The tyrosine phosphatase Shp2 in development and
cancer. Adv Cancer Res. 106:53–89. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mohi MG and Neel BG: The role of Shp2
(PTPN11) in cancer. Curr Opin Genet Dev. 17:23–30. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bard-Chapeau EA, Li S, Ding J, Zhang SS,
Zhu HH, Princen F, Fang DD, Han T, Bailly-Maitre B, Poli V, et al:
Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular
carcinogenesis. Cancer Cell. 19:629–639. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Jiang C, Hu F, Tai Y, Du J, Mao B, Yuan Z,
Wang Y and Wei L: The tumor suppressor role of Src homology
phosphotyrosine phosphatase 2 in hepatocellular carcinoma. J Cancer
Res Clin Oncol. 138:637–646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Chan TA and Heguy A: The protein tyrosine
phosphatase receptor D, a broadly inactivated tumor suppressor
regulating STAT function. Cell Cycle. 8:3063–3064. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ostman A, Hellberg C and Bohmer FD:
Protein-tyrosine phosphatases and cancer. Nat Rev Cancer.
6:307–320. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
McFarland BC and Benveniste EN: Reactive
astrocytes foster brain metastases via STAT3 signaling. Ann Transl
Med. 7 (Suppl 3):S832019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Chen J, Jiang CC, Jin L and Zhang XD:
Regulation of PD-L1: A novel role of pro-survival signalling in
cancer. Ann Oncol. 27:409–416. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yang L, Sun YY, Liu YR, Yin NN, Bu FT, Yu
HX, Du XS, Li J and Huang C: PTP1B promotes macrophage activation
by regulating the NF-κB pathway in alcoholic liver injury. Toxicol
Lett. 319:11–21. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Chong ZZ and Maiese K: The Src homology 2
domain tyrosine phosphatases SHP-1 and SHP-2: Diversified control
of cell growth, inflammation, and injury. Histol Histopathol.
22:1251–1267. 2007.PubMed/NCBI
|
|
80
|
Han T, Xiang DM, Sun W, Liu N, Sun HL, Wen
W, Shen WF, Wang RY, Chen C, Wang X, et al: PTPN11/Shp2
overexpression enhances liver cancer progression and predicts poor
prognosis of patients. J Hepatol. 63:651–660. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
An H, Hou J, Zhou J, Zhao W, Xu H, Zheng
Y, Yu Y, Liu S and Cao X: Phosphatase SHP-1 promotes TLR- and
RIG-I-activated production of type I interferon by inhibiting the
kinase IRAK1. Nat Immunol. 9:542–550. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Tai WT, Shiau CW, Chen PJ, Chu PY, Huang
HP, Liu CY, Huang JW and Chen KF: Discovery of novel Src homology
region 2 domain-containing phosphatase 1 agonists from sorafenib
for the treatment of hepatocellular carcinoma. Hepatology.
59:190–201. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Chen KF, Tai WT, Hsu CY, Huang JW, Liu CY,
Chen PJ, Kim I and Shiau CW: Blockade of STAT3 activation by
sorafenib derivatives through enhancing SHP-1 phosphatase activity.
Eur J Med Chem. 55:220–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Fan LC, Shiau CW, Tai WT, Hung MH, Chu PY,
Hsieh FS, Lin H, Yu HC and Chen KF: SHP-1 is a negative regulator
of epithelial-mesenchymal transition in hepatocellular carcinoma.
Oncogene. 34:5252–5263. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wen LZ, Ding K, Wang ZR, Ding CH, Lei SJ,
Liu JP, Yin C, Hu PF, Ding J, Chen WS, et al: SHP-1 acts as a tumor
suppressor in hepatocarcinogenesis and HCC progression. Cancer Res.
78:4680–4691. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Su JC, Tseng PH, Hsu CY, Tai WT, Huang JW,
Ko CH, Lin MW, Liu CY, Chen KF and Shiau CW: RFX1-dependent
activation of SHP-1 induces autophagy by a novel obatoclax
derivative in hepatocellular carcinoma cells. Oncotarget.
5:4909–4919. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Qian H, Deng X, Huang ZW, Wei J, Ding CH,
Feng RX, Zeng X, Chen YX, Ding J, Qiu L, et al: An HNF1α-regulated
feedback circuit modulates hepatic fibrogenesis via the crosstalk
between hepatocytes and hepatic stellate cells. Cell Res.
25:930–945. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zhang J, Li Z, Liu L, Wang Q, Li S, Chen
D, Hu Z, Yu T, Ding J, Li J, et al: Long noncoding RNA TSLNC8 is a
tumor suppressor that inactivates the interleukin-6/STAT3 signaling
pathway. Hepatology. 67:171–187. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Duran A, Hernandez ED, Reina-Campos M,
Castilla EA, Subramaniam S, Raghunandan S, Roberts LR, Kisseleva T,
Karin M, Diaz-Meco MT and Moscat J: p62/SQSTM1 by binding to
vitamin D receptor inhibits hepatic stellate cell activity,
fibrosis, and liver cancer. Cancer Cell. 30:595–609. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ,
Tao QF, Liu F, Pan W, Wang TT, Zhou CC, et al: A long noncoding RNA
activated by TGF-β promotes the invasion-metastasis cascade in
hepatocellular carcinoma. Cancer Cell. 25:666–681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Li N, Zhou ZS, Shen Y, Xu J, Miao HH,
Xiong Y, Xu F, Li BL, Luo J and Song BL: Inhibition of the sterol
regulatory element-binding protein pathway suppresses
hepatocellular carcinoma by repressing inflammation in mice.
Hepatology. 65:1936–1947. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zhang L, Yang Z, Ma A, Qu Y, Xia S, Xu D,
Ge C, Qiu B, Xia Q, Li J and Liu Y: Growth arrest and DNA damage
45G down-regulation contributes to Janus kinase/signal transducer
and activator of transcription 3 activation and cellular senescence
evasion in hepatocellular carcinoma. Hepatology. 59:178–189. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Gough DJ, Corlett A, Schlessinger K,
Wegrzyn J, Larner AC and Levy DE: Mitochondrial STAT3 supports
Ras-dependent oncogenic transformation. Science. 324:1713–1716.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wang YQ, Zhang F, Tian R, Ji W, Zhou Y,
Sun XM, Liu Y, Wang ZY and Niu RF: Tyrosine 23 phosphorylation of
annexin A2 promotes proliferation, invasion, and Stat3
phosphorylation in the nucleus of human breast cancer SK-BR-3
Cells. Cancer Biol Med. 9:248–253. 2012.PubMed/NCBI
|
|
95
|
Yamada S, Shiono S, Joo A and Yoshimura A:
Control mechanism of JAK/STAT signal transduction pathway. FEBS
Lett. 534:190–196. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhang Z, Shen K, Lu W and Cole PA: The
role of C-terminal tyrosine phosphorylation in the regulation of
SHP-1 explored via expressed protein ligation. J Biol Chem.
278:4668–4674. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Xiang D, Cheng Z, Liu H, Wang X, Han T,
Sun W, Li X, Yang W, Chen C, Xia M, et al: Shp2 promotes liver
cancer stem cell expansion by augmenting beta-catenin signaling and
predicts chemotherapeutic response of patients. Hepatology.
65:1566–1580. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Chen YN, LaMarche MJ, Chan HM, Fekkes P,
Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH, Chen Z, Cooke
VG, et al: Allosteric inhibition of SHP2 phosphatase inhibits
cancers driven by receptor tyrosine kinases. Nature. 535:148–152.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Leung CON, Tong M, Chung KPS, Zhou L, Che
N, Tang KH, Ding J, Lau EYT, Ng IOL, Ma S and Lee TKW: Overriding
adaptive resistance to sorafenib through combination therapy with
Src homology 2 domain-containing phosphatase 2 blockade in
hepatocellular carcinoma. Hepatology. 72:155–168. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Kang HJ, Chung DH, Sung CO, Yoo SH, Yu E,
Kim N, Lee SH, Song JY, Kim CJ and Choi J: SHP2 is induced by the
HBx-NF-κB pathway and contributes to fibrosis during human early
hepatocellular carcinoma development. Oncotarget. 8:27263–27276.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Lo J, Lau EY, Ching RH, Cheng BY, Ma MK,
Ng IO and Lee TK: Nuclear factor kappa B-mediated CD47
up-regulation promotes sorafenib resistance and its blockade
synergizes the effect of sorafenib in hepatocellular carcinoma in
mice. Hepatology. 62:534–545. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Mohi MG, Williams IR, Dearolf CR, Chan G,
Kutok JL, Cohen S, Morgan K, Boulton C, Shigematsu H, Keilhack H,
et al: Prognostic, therapeutic, and mechanistic implications of a
mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer
Cell. 7:179–191. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zhang SQ, Yang W, Kontaridis MI, Bivona
TG, Wen G, Araki T, Luo J, Thompson JA, Schraven BL, Philips MR and
Neel BG: Shp2 regulates SRC family kinase activity and Ras/Erk
activation by controlling Csk recruitment. Mol Cell. 13:341–355.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Yang X, Tang C, Luo H, Wang H and Zhou X:
Shp2 confers cisplatin resistance in small cell lung cancer via an
AKT-mediated increase in CA916798. Oncotarget. 8:23664–23674. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ran H, Tsutsumi R, Araki T and Neel BG:
Sticking it to cancer with molecular glue for SHP2. Cancer Cell.
30:194–196. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Wu C, Sun M, Liu L and Zhou GW: The
function of the protein tyrosine phosphatase SHP-1 in cancer. Gene.
306:1–12. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Barr AJ, Ugochukwu E, Lee WH, King ON,
Filippakopoulos P, Alfano I, Savitsky P, Burgess-Brown NA, Müller S
and Knapp S: Large-scale structural analysis of the classical human
protein tyrosine phosphatome. Cell. 136:352–363. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Tai WT, Hung MH, Chu PY, Chen YL, Chen LJ,
Tsai MH, Chen MH, Shiau CW, Boo YP and Chen KF: SH2
domain-containing phosphatase 1 regulates pyruvate kinase M2 in
hepatocellular carcinoma. Oncotarget. 7:22193–22205. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Pfirsch-Maisonnas S, Aloulou M, Xu T,
Claver J, Kanamaru Y, Tiwari M, Launay P, Monteiro RC and Blank U:
Inhibitory ITAM signaling traps activating receptors with the
phosphatase SHP-1 to form polarized ‘inhibisome’ clusters. Sci
Signal. 4:ra242011. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Alsadeq A, Hobeika E, Medgyesi D, Klasener
K and Reth M: The role of the Syk/Shp-1 kinase-phosphatase
equilibrium in B cell development and signaling. J Immunol.
193:268–276. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Viant C, Fenis A, Chicanne G, Payrastre B,
Ugolini S and Vivier E: SHP-1-mediated inhibitory signals promote
responsiveness and anti-tumour functions of natural killer cells.
Nat Commun. 5:51082014. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Motiwala T, Kutay H, Ghoshal K, Bai S,
Seimiya H, Tsuruo T, Suster S, Morrison C and Jacob ST: Protein
tyrosine phosphatase receptor-type O (PTPRO) exhibits
characteristics of a candidate tumor suppressor in human lung
cancer. Proc Natl Acad Sci USA. 101:13844–13849. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Motiwala T, Ghoshal K, Das A, Majumder S,
Weichenhan D, Wu YZ, Holman K, James SJ, Jacob ST and Plass C:
Suppression of the protein tyrosine phosphatase receptor type O
gene (PTPRO) by methylation in hepatocellular carcinomas. Oncogene.
22:6319–6331. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Zhang W, Hou J, Wang X, Jiang R, Yin Y, Ji
J, Deng L, Huang X, Wang K and Sun B: PTPRO-mediated autophagy
prevents hepatosteatosis and tumorigenesis. Oncotarget.
6:9420–9433. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Mareninova OA, Hermann K, French SW,
O'Konski MS, Pandol SJ, Webster P, Erickson AH, Katunuma N,
Gorelick FS, Gukovsky I and Gukovskaya AS: Impaired autophagic flux
mediates acinar cell vacuole formation and trypsinogen activation
in rodent models of acute pancreatitis. J Clin Invest.
119:3340–3355. 2009.PubMed/NCBI
|
|
117
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|